Introduction
Cancer that occurs in families more often than would be expected by chance is termed as familial cancers [1]. These rare cancers often occur at an early age. FCS increase the risk of cancers and are associated with various physical symptoms. They may also be a sign of shared environmental or lifestyle factors that resulted in inherited genetic mutation [2,3].
The following clues provided by Neel and Schull (1954) categorize a tumour to be of genetic origin:
The occurrence of the disease in defined proportions among persons related by descent, when environmental causes can be ruled out.
The failure of the disease to appear in unrelated lines (e.g., in spouses or in-laws)
A characteristic age of onset and course, in the absence of known precipitating factors.
Greater concordance in monozygotic than in dizygotic twins [4].
A clinician should be cautious and suspect a FCS when two or more members of the same side of a family present with similar kind of cancer, an earlier age of onset, has birth defects, when a person has more than one primary cancer, family member with cancer clusters belonging to known FCS and with an evidence of inherited gene mutation [5].
The inheritance pattern seen in familial cancer syndromes is presented in [Table/Fig-1] [2,6].
Inheritance pattern in familial syndromes.
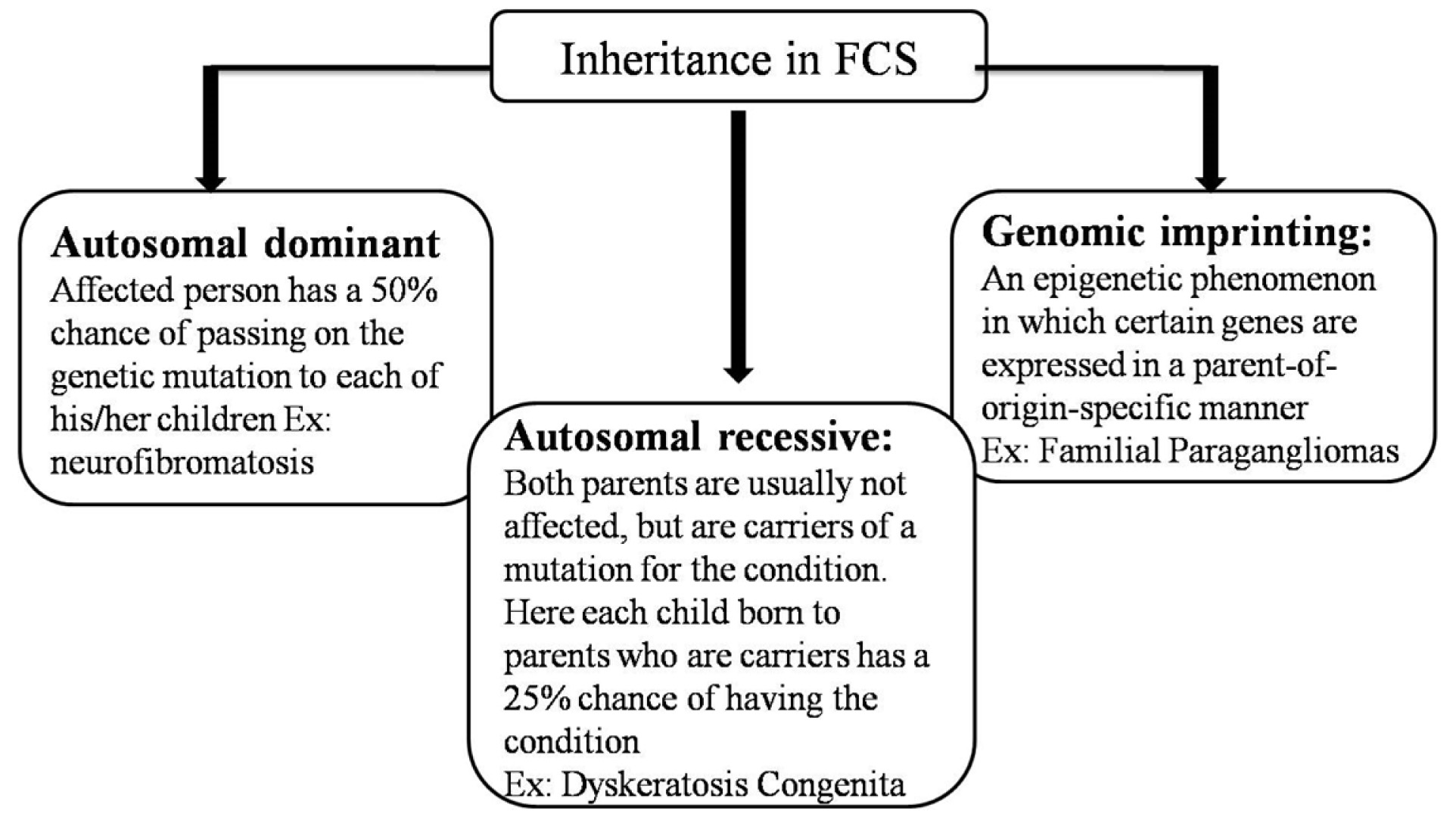
The primary genetic event either a germline mutation in a tumour suppressor or an oncogene has been identified in approximately 19 FCS. Patients predisposed to FCS inherit one mutant allele of genes (either maternal or paternal) and neoplasia develops only when additional mutation of remaining allele occurs. For instance, in syndromes of tumour suppressor or caretaker genes, patients inherit a single mutant allele of a caretaker gene from an affected parent and only develop cancer after three further somatic mutations (the remaining allele of the caretaker gene and both alleles of a gatekeeper gene). In contrast, sporadic tumours develop in individuals who do not have a germline mutation but who acquire, over a period of time, somatic mutations of both alleles. In general, germline mutations are scattered largely along the entirety of the gene [1,2,7].
Familial cancers often involve multiple organ systems and affect paired organs with bilateral or multifocal tumours. Affected individuals can have more than one type of cancer and malignancies other than epithelial are more likely to occur (soft tissue sarcomas, leukaemias/ lymphomas, nervous system tumours) [8].
A search was conducted with keywords “familial tumours”, “familial cancer syndromes” and “familial tumours in head and neck” in Pubmed, Pubmed Central, Science Direct, Wiley Interscience, Google, Google Scholar to identify the FCS associated with head and neck region. Cancers with predominant oral origin and manifestations were mainly considered.
The familial cancers thus identified are listed in [Table/Fig-2] [2,3,9-14]:
Familial cancers with oral manifestations.
| Sl No. | Systems associated | Neoplasms |
|---|
| 1. | Endocrine system associated | 1.Multiple endocrine neoplasia syndrome2.Familial hyperparathyroidism with jaw tumour syndrome |
| 2. | Carcinomas | 1.1.Oral Squamous Cell Carcinomas (OSCC)2.Nasopharyngeal carcinomas3.Familial Malignant melanoma4.Familial Oncocytosis in salivary glands |
| 3. | Soft tissue tumours | 1.Paragangliomas2.Neurofibromatosis |
| 4. | Other Associated Syndromes | 1.Muir Torr syndrome2.Gorlin-Goltz syndrome.3.Li-Fremauni syndrome.4.Cowden’s syndrome.5.Gardner’s syndrome.6.Peutz-Jeghers syndrome7.Familial adenomatosis polyposis |
Multiple Endocrine Neoplasia Syndrome
Multiple Endocrine Neoplasia (MEN) syndrome, an autosomal dominant disorder is characterized by simultaneous occurrence of tumours involving two or more endocrine glands derived from neural crest. They develop tumours involving parathyroid gland, pancreas, pituitary gland, adrenal gland and thyroid gland [15].
The major forms of MEN are MEN 1 or Wermer’s syndrome with menin (protein product of MEN 1 gene) mutations, MEN2/MEN2A/ Sipple’s syndrome with mutations in Rearranged During Transfection (RET) protoncogene a transmembrane tyrosine kinase receptor, MEN3/ MEN2B/ Wagenmann-Froboese/mucosal neuroma syndrome with RET mutations located on 10q11.2 and MEN4/MEN X with cyclin-dependent kinase inhibitor (CDNKI) B mutations [15,16].
The oral manifestations are most prominent in MEN 2B and hence the manifestations of this syndrome are discussed [17-19].
The incidence rate is 1 in 30,000. Approximately 95% of the MEN 2B patients carry M918T (exon 16) missense mutation, whereas others harbor a mutation at codon 883 and 918 (exon 15) in the tyrosine kinase domain. These mutations interfere with ATP binding, altering the substrate specificity of RET substrate recognition pocket which leads to ligand-independent activation or modification of kinase activity [16].
Pathological mucosal nerve proliferations associated with increase in size and number of nerve fascicle with thickened perineurium is seen. No intradermal axon flare response to histamine is noticed. Intraosseous neural hyperplasia is a specific finding of the MEN 2B syndrome. The oral mucosal lesions are more commonly neurofibromas (plexiform type) than neuromas [17-19].
Around 25% of medually thyroid carcinoma are hereditary, associated with MEN 2 syndrome and present with early aggressive behaviour. They are bilateral and metastasize early [15].
Familial Hyperparathyroidism with Jaw Tumour Syndrome
Primary hyperparathyroidism results from persistent hypercalcaemia with inadequate secretion of parathyroid hormone. About 5%-10% of cases are inherited (autosomal dominant trait) as a part of syndromes such as multiple endocrine neoplasia, Hyperparathyroidism Jaw-Tumour (HPT-JT), Neonatal Severe Hyperparathyroidism (NSHPT), Familial Hypocalciuric Hypercalcaemia (FHH), Familial Isolated Hyperparathyroidism (FIHPT) and Autosomal Dominant Moderate Hyperparathyroidism (ADMH) and most other cases are sporadic [9,20].
Familial types are more heterogenous, presents with multiglandular parathyroid disease (conventionally parathyroid hyperplasia) and occur with equal frequencies in both sexes [9,20].
HPT-JT presents with oral manifestations and around 300 cases from approximately 100 families (world-wide) have been described till date. Virtually all patients with HPT-JT develop primary hyperparathyroidism and average age of onset is 32 years [9,20,21].
Inactivating mutations of cell division cycle 73 (CDC73)/hyperparathyroidism 2 (HRPT2) gene (1q31.2) that encodes parafibromin, a tumour suppressor protein in parathyroid gland is noticed. It interacts with Polymerase Associated Factor 1 (PAF1) to regulate histone ubiquitination, apoptosis, gene transcription, growth factor gene transcription, cyclin D1 signalling and Wnt/b-catenin signalling. Bi-allelic inactivation of CDC73/HRPT2 and complete loss of parafibromin expression results in parathyroid cancer [9].
Severe hypercalcaemia (albumin corrected calcium >3 mmol/l), a large palpable neck mass of size more than 3 cm, third/second generation PTH assay ratio >1, infiltrative parathyroid gland disease or suspected metastasis on sestamibi scintigraphy are all the features of parathyroid cancer [9].
In patients aged less than 35 years with hyperparathyroidism, parathyroid cancer/cystic change, loss of parafibromin staining or family history of HPT-JT syndrome; genetic analysis for HPT-JT should be performed [9,20,21].
Familial Oral Squamous Cell Carcinoma
Oral squamous cell carcinoma is a malignant epithelial tumour with squamous differentiation characterized by the formation of keratin or the presence of intercellular bridges or both [22].
Familial OSCC occurs as an autosomal dominant disorder and the estimated risks of first degree relatives have been reported to be 1.1, 9.25, 103.5 or 3.812 by various studies. The homozygous genotype may increase the risk/odds ratio to 1.77. It can be acquired as a genetic trait in families with high-risk habits such as smoking and alcohol use [22,23]. Evidence for hereditary syndrome associated OSCC includes occurrence of tumours in family groups having different lifestyles, early onset in non-smokers or individuals lacking any risk factors, and/or aggregation in families by tumour site. Development of second primary tumours is the main cause of treatment failure [23].
The following criteria was selected in one of the study for familial OSCCs: 1) minimum of two first-degree relatives affected with head and neck cancer and/or other related cancers (cancer of stomach, kidney, breast, uterus, cervix etc.,); 2) less than 45 years of age of onset in at least one of the affected family members; 3) OSCC patient with no known aetiological factors such as tobacco or alcohol use, at any age of onset [24].
Mutation of various genes is observed, such as germ line p53 alterations with first degree relatives affected by multiple malignancies, missense mutations located primarily in exons 4+/- 9 with hot spot codons 205+/-248 associated with Li Fraumeni syndrome and impairment in regulator proteins such as overexpression of MDM2. Overexpression of MDM2 is associated with 52% increase in premalignant lesions and 78% increase in OSCC in Indians [7]. The CDKN 2 locus is frequently deleted; a protein encoded by the ADP-Ribosylation Factor 1 (ARF) from this locus binds mouse double minute 2 (MDM2) and prevents p53 degradation. Loss of ARF, thus leads to a reduction in wild-type p53 levels resulting in a reduced p53 response to DNA damage [7].
Germline TP16 mutations are associated with increased p16/ CDKN2A protein levels that accumulate in senescent oral keratinocytes resulting in keratinocyte immortalisation. Inactivation of p16/CDKN2A can occur by mutation, methylation of promoter sequences or by gene deletion and has been reported in approximately 80% of OSCCs [7].
The results of a study done by Bertonhaa FB et al., suggested that genetic and epigenetic changes involved in loss of function of the PHD (cys 4-his-cys 3 motif in plant homeodomain) finger protein 21B (PHF21B) gene that encodes for PHD finger protein 21B (nuclear protein) of 531 amino acids is observed in OSCC. PHF21B acts as a tumour suppressor as its expression reduces cell migration and colony formation [24].
Polymorphisms of Glutathione-S-Transferese P (GSTP) 1 that encodes a cytoplasmic protein involved in the detoxification of carcinogenic metabolites may be associated with susceptibility to oral cancer [7].
Patients with xeroderma pigmentosum, Fanconi’s anemia and Dyskeratosis Congenita (DC) present with an early onset of OSCC. Two major severe subsets of DC include Hoyeraal-Hreidarsson syndrome and Revesz syndrome [7,25-27].
DC or Zinsser-Engman-Cole syndrome is characterized by mutations in the components of the telomere maintenance machinery resulting in excessively short telomeres especially in highly proliferating tissues [25,26]. These mutations especially DKC1 and TIN 2 results in short telomeres. DKC1 gene encodes a protein Dyskerin which is an essential component of telomerase complex. TINF2 encodes protein TIN2, a component of Shelterin which binds to telomere DNA repeats, protecting them from degradation [26,27].
Through successive generations, the telomeres become critically short and affect the ability of stem cells to continually renew tissues that possess high cellular turn over. Dysfunctional telomeres cause early loss of progenitor cells of rapidly dividing tissues. Loss of dividing progenitor cells starts a vicious cycle by increasing the number of cells recruited into cell cycles. Dysfunctional telomeres activate p53 pathway and increase genomic stability leading eventually to cell crisis, cell cycle arrest, senescence and cell death. Potentially malignant cells develop when rare cells escape cell senescence death and acquire a mechanism to maintain telomere integrity [26].
The other syndromes associated with OSCC include Bloom’s syndrome, Lynch-II syndrome, and Ataxia-telangiectasia [22].
Nasopharyngeal Carcinoma
Familial Nasopharyngeal Carcinoma (NPC) occurs in at least two first degree relatives and has striking ethnic and geographic distribution. It is the leading cause of death in Cantonese and Chinese population. The aetiology of NPC is multifactorial, interaction of race, genetics, environment and Epstein-Barr virus. Reports of genetic clusters suggest a genetic component. Involvement of dietary factors such as consumption of nitrosamine-rich salted fish as seen in South East Asia have one of the highest incidences of NPC [22,28].
NPC is associated with chromosomes 3p21.31-21.2, 4p15.1-q12 and results in poorly differentiated tumours [8].Genetic polymorphisms of some metabolic enzyme genes Cytochrome p450 family 2 sub family E member 1 (CYP2E1) and Glutathione-S-Transferase Mu 1 (GSTM1) and DNA repair genes such as X-ray Repair Cross Complementing 1 (XRCC1) and human Oxoguanineglycosylase 1 (hOGG1) influence susceptibility to NPC. Certain Human Leukocyte Antigen (HLA) groups were also reported to be associated with increased NPC risk [29].
Most patients presents with stage III or IV disease and are aged between 31 to 50 years.
According to a study by Loh KS et al., sex, age and race were similar in familial and non-familial NPC and there were no differences in symptoms between the two groups [30].
Familial Malignant Melanoma
Familial melanoma occurs in the setting of unilateral lineage, with a complex phenotype of early age of onset, multiple atypical moles, multiple primary melanomas, multiple melanomas in the family and in some instances pancreatic cancer [31].
A family history of melanoma is seen in about 10% of cases with an increased risk raise from 4 to >1000 fold; however, individuals with features of true hereditary melanoma are rare. A germline mutation of CDKN2A should be suspected if multiple atypical nevi are found in presence of familial melanomas [31]. CDKN2A chromosomal locus encodes inhibitor p16INK4a (or p16) which is a CDK4/6 inhibitor that induces cellular senescence and helps in DNA repair before mitoses. The p16 regulates intracellular oxidative stress in a cell cycle-independent manner and may also mediate additional functions as it is observed that in vitro, familial melanoma associated p16 mutants still retain CDK4 binding capacity. Incidence of melanoma in p16 mutant relatives is highly associated with chronic exposure to UV radiation due to production of Reactive Oxygen Species (ROS) in the skin. As compared to keratinocytes and fibroblasts, melanocytes are highly susceptible to oxidative stress due to p16 mutation. Hence, it can be concluded that melanocytes are dependent more on p16 for normal oxidative regulation than other cell types which reasons partially on increased incidence of melanoma over other cancers in p16 mutants [32].
Additional mutations seen include CDK4, germline variants in TERT, MITF and BAP1. Melanocytic BAP1-mutation presents with atypical intradermal tumours which should be recognized and treated early [33].
Paraganglioma
The paraganglioma (PG) are defined as neuroendocrine tumours arising from paraganglia and collections of neural crest-derived paraganglionic neuroectodermal cells present throughout the body outside the adrenal system [34,35].
These neoplasms comprise 0.03% of all tumours and 0.6% of all head and neck tumours [34]. Autosomal dominant mode of inheritance with complete penetrance and recently genomic imprinting has been associated with familial PG [36].
In head and neck region, PG arise in 1) carotid body (50%-70%) termed carotid body tumours (CBT) 2) vagus nerve sheath ganglions (5%), 3) chief cells of jugular bulb and 4) along auricular nerve within the tympanic space (20-40%). Head and Neck PG (HNPGLs) are parasympathetically derived and only rarely secrete catecholamines contrasting their sympathetic abdominal counterpart [34].
The syndromes associated with CBTs typically originate in germline mutations of the A, B, C, and D subunits of the mitochondrial complex II enzyme Succinate Dehydrogenase (SDH). SDH catalyses the oxidation of succinate to fumarate in the Krebs cycle and it is involved in the oxidative phosphorylation system where it acts as an electron donor. This mechanism plays a critical role in oxygen sensing and signalling capabilities. Inactivation of SDH due to heterozygous germline mutations would probably mimic a chronic hypoxic stimulation of the chief paraganglionic cells resulting in increased levels of downstream hypoxia effector molecules, leading to cellular proliferation, hyperplasia and ultimately neoplastic transformation of carotid body cells. Genetic markers of hypoxia such as Hypoxia Inducible Factor (HIF)-1 and HIF-2 are upregulated in familial PG syndromes [34].
SDHB carriers (10%-20%) who are young have an increased incidence of head and neck PG malignancy (6-24%, common in carotid body) and abdominal paragangliomas (abPG). It is associated with increased urine metanephrines and plasma catecholamines mainly dopamine levels. Malignant PGs have been known to express biochemically less mature breakdown products [34,35].
SDHD mutations in head and neck PG are identified in 50-94% of cases. They predispose to multifocal PHEOs/PGLs, primarily from the head and neck region, with low metastatic risk [37]. Plasma levels of dopamine and methoxytyramine higher than 0.2 nmol/l constitutes a useful biomarker to detect metastases in such cases [35].
Neurofibromatosis
Neurofibromatosis (NF) is a group of autosomal dominant genetic disorders characterized by multiple cutaneous lesions and tumours of the central and peripheral nervous system. NF is categorized into 2 types NF1 and NF 2 [37].
Neurofibromatosis 1
NF 1 or von Recklinghausen’s disease presents with an incidence of 1 per 2500–3000 individuals and an overall prevalence of 1 in 5000 [38].
NF1 is a familial disorder with a mendelian inheritance pattern, with half of cases caused by newly appearing mutations. It is caused due to a defect in gene located in pericentric region of chromosome 17 on 17q11.2, a multifunctional tumour suppressor gene coding for protein neurofibromin. The development of the disease requires Loss of Heterozygosity (LOH) involving only one cell type in each case that subsequently expands clonally to form a visible lesion. Majority of NF 1 mutations occur due to point changes in the gene coding sequences whereas gene deletion occurs only in a minority of cases. There are mosaic forms in which a late somatic mutation may give rise to mosaic NF. If the mosaicism is limited to one or several body segments it results in segmental or localized NF, whereas early mutations gives rise to conditions clinically indistinguishable from the generalized forms. Type 2 mosacisim is a condition in which a more affected segment is superimposed on generalized disease [39,40].
Loss of function of NF 1 gene results in a RAS opathy. The protein neurofibromin inhibits proliferative growth through blockade of RAS-mediated signal transduction. RAT sarcoma viral oncogene homologue (RAS) is implicated in developmental process such as cell proliferation, differentiation, organogenesis, synaptic plasticity, growth, apoptosis and cell ageing. The protein neurofibromin can also be considered as a modulator of cell motility and cell adhesion. It acts as a negative regulator of rhokinase/rho-associated protein kinase (RHO/ROCK) signalling pathways involved in cytoskeletal dynamics that are instrumental in proper neuronal development [38]. Neurofibromin is expressed in Schwann cells, melanocytes, leukocytes, adrenal glands, neurons and astrocytes of central nervous tissues [40].
Along with criteria put forth by National Institute of Health (NIH), recently, suggestions have been made to add few clinical signs to the diagnostic criteria and include:
Genetic analysis of NF 1 gene.
Cutaneous signs: Nevus Anemicus, juvenile xanthogranuloma, mixed vascular hamartomas, soft-touch skin and hyperpig mentation.
Extracutaneous signs: Astrocytic hamartomas or angiomas; choroidal hamartomas; large head circumference; hypertelorism; unidentified bright objects which are benign progressive lesions in cerebrum; learning, speech and behavioural disabilities; headache and seizures; and neoplasms such as rhabdomyosarcoma, leukaemia, lymphomas, Malignant Peripheral Nerve Sheath Tumours (MPNST), gastrointestinal tumours (4-25%) and pheochromocytomas (0.1% to 5%) [39-41].
The oral manifestations of NF 1 are seen in 72% of cases [37, 38, 42].
Neurofibromatosis 2
A rare disorder with the incidence of 1 in 25,000 to 1 in 40,000 caused due to mutation in NF2 gene located on chromosome 22 (22q12). NF2 gene is a tumour suppressor gene and its product merlin binds to actin or CD44 molecules which are involved with cell interaction with extracellular matrix. The criteria for diagnosis are presented in [Table/Fig-3] [43].
Familial syndromes with oral stigmata.
| Sl No | Hereditary syndromes with oral stigmata | Gene mutation | General manifestations | Oral manifestations |
|---|
| 1. | MEN 2B | RET | Coarse facies, apertognathia, and marfanoid habitus, wide eye expression | Facial asymmetry, nodular and diffuse enlargement of lips, hypertrophic tongue and mucosal neuromas on buccal, lingual and labial mucosa.Shortened mandibular incisor and molar roots. Central diastema, widened and bifurcated mandibular canal [17-19]. |
| 2. | HPT-JT | HRPT2 gene | 15% of cases develop parathyroid cancer. Wilms’ tumour, Polycystic disease, tumours of kidney, primary hyperparathyroidism and uterine cysts/hamartomas | 30% of cases show fibro-osseous lesions of maxilla and mandible mainly ossifying fibromas rather than brown tumours with giant cells [22]. |
| 3. | Familial OSCC:a) Xeroderma Pigmentosum | Impairment of nucleotide excision repair mechanism | Carcinoma skin, actinic keratosis, pigmentation and progressive neurological degeneration [7] | Oral keratinocytes infected with HPV 16. Carcinoma tongue: incidence is 10,000 times the normal [7] |
| b) Fanconi anemia | Autosomal recessive genomic instability | Bone marrow failure, leukaemia, congenital defects [7] | OSCC |
| c) Dyskeratosis congenital [26,27,28] | Mutations in DKC1 (40%), TERC, TERT, TINF2 (10%), NOPIO, NHP2, TCAB1, C16, CTC1, and RTEL1. X-linked recessive, autosomal dominant or recessive. | Inherited bone marrow failure 3 pathognomonic features:a)Dystrophic nailsb)Oral leukoplakia (80%) and 30% progress to malignancyc)Abnormal skin pigmentation | Leukoplakia on tongue, buccal mucosa, palate. Periodontal disease, hyperpigmentation of buccal mucosa, enamel hypocalcification, taurodontism. Skeletal abnormalities (20%): mandibular hypoplasia, abnormal trabeculation, and avascular necrosis. |
| 4. | Neurofibromatosis 1 | NF 1 gene; Generalized increase in osteoclastic activity. | NIH,1988 diagnostic criteria: cafe-au-lait macules, freckling, two or more neurofibromas or one plexiform neurofibroma, Lich nodules, optic gliomas, bone dysplasias and first degree relatives with NF1 | Fibrous gingival enlargements, pigmentations, periodontal manifestations, enamel hypoplasia. Neurofibromas involving the tongue, palate, vestibule, buccal mucosa, lip, floor of the mouth, gingiva (5%), macroglossia (plexiform neurofibroma) and enlargement of fungiform papillae.Osseous manifestations: wide inferior alveolar canals and enlarged mandibular foramen. Mandibular deformities of the coronoid notch pathognomonic sign of NF [37,38,42]. |
| 5. | Neurofibromatosis 2 | NF 2 gene | Diagnostic criteria according to National Institute of Health (NIH): Bilateral eight nerve masses or A first degree relative with NF 2 and with a unilateral eight nerve mass or two of the following: a) neurofibroma; b) meningioma; c) glioma; d) schwannoma; or e) juvenile posterior subcapsular cataract [43] | Neurofibromas, Schwanommas |
| 6. | Neurofibromatosis 3 | Germline mosaicism of SMARCB1 | Molecular diagnosis:1.2 or more schwanommas or meningiomas and genetic studies of at least 2 tumours showing LOH of chromosome 22 and SMARC1 mutation.2.One schwannoma or meningioma and a germline pathogenic SMARCB1 mutation. Clinical diagnosis:1.2 or more non-intradermal schwannomas and absence of vestibular schwannoma.2.One schwannoma or meningioma and affected first degree relative.3.Possible diagnosis when two or more non-intradermal schwannomas and chronic pain associated with tumour exists.4.Absence of ocular pathology, ependymomas and vestibular schwannomas [43,44]. | Schwanommas |
| 7. | Muir Torr syndrome (MTS) | Mismatch repair genes: MLH1, MSH2, MSH6. | Three family members or one first degree relative to two other members with visceral carcinoma such as sebaceous tumour, keratoacanthomas as defined by Amsterdam criteria. | Keratoacanthomas, vascular aberrations in the oral mucosa, Fordyce granules |
| 8. | Gorlin-Goltz syndrome. | 9q22.3-9q31, PTCH1 gene | Multiple basal cell carcinomas, hyperkeratosis of palms and soles. bi-fid rib (2nd and 7th), macrocephaly, intracranial ectopic calcifications, cardiac tumours, ovarian fibromas, fibro-sarcomas, foetal rhabdomyomas | Odontogenic keratocyst, ameloblastoma, impacted teeth, dental agenesis, mandibular prognathism, hyperplasia of the mandibular coronoid processes |
| 9. | Li-Fremauni syndrome | TP53 | Osteosarcomas, leukaemia, malignant neuroepithelial tumours and primitive neuroectodermal tumours | OSCC |
| 10. | Cowden’s syndrome | Chromosome 10q22-23, PTEN/ MMAC1 gene;. | Major criteria: mammary carcinoma, follicular thyroid carcinoma macrocephaly, Lhermitte-Duclos disease and endometrial carcinoma.Minor criteria: thyroid disorders, mental retardation, intestinal hamartomatous polyps, fibrocystic mammary disease, lipomas, hemangiomas, ganglioneuromas fibromas, and urogenital malformations | Facial trichilemmomas, small verrucous papules on the gums and lips in 80% cases. Papular fibromas, scrotal tongue, ogival palate, periodontal disease, adenoid facies. |
| 11. | Gardner’s syndrome | Adenomatosis polyposis (APC) | Gastrointestinal polyps, desmoid tumours, epidermoid cysts, soft tissue tumours | Osteomas of the skull and mandible tooth impaction, odontomas and cementomas, congenitally missing teeth, supernumerary teeth, dentigerous cysts, hypodontia, complete absence of the periodontal space caused by hypercementosis |
| 12. | Familial adenomatosis polyposis | Adenomatosis polyposis (APC) gene | Adenomatous polyps in colorectum with 100% risk of malignant transformation epidermoid cysts osteomas, desmoids tumours, papillary thryroid carcinoma and adrenocortical tumours | Oral manifestations :>70% supernumerary teeth, dentigerous cysts, congenitally missing teeth, fused roots of molars. |
| 13. | Peutz-Jeghers syndrome | STK11/LKB1 gene on 19p13.3 | Hamartomatous polyps of the gastrointestinal tract with increased malignancy risk. | Melanin spots freckling which are "periorificial," clustered around the mouth, eyes, nostrils, and perianal area. |
Neurofibromatosis 3
Previously considered as a subtype of NF2; schwannomatosis or NF 3 is a relatively new condition. It is diagnosed when multiple schwannomas without eighth nerve involvement is noticed. Family history is present in 15-25% of cases. Germline mosaicism of SMARCB1 (INI-1) gene localized on chromosome 22 near NF2 gene is seen. The diagnostic criteria are presented in [Table/Fig-3] [43].
Exclusion Criteria
Germiline pathogenic NF2 mutation.
Fulfilled criteria for NF2.
First degree relative with NF2.
Schwannomas in radiation field only [44].
Familial Oncocytosis
Oncocytes are large epithelial cells with an eosinophilic granular cytoplasm containing many mitochondria (60%) with a swollen appearance. These mitochondria show both structural (globular with lamelliform tubular, or flat cristae) and functional alteration. Familial oncocytic tumours mainly occurs when there is a defect in gene coding mitochondrial respiratory enzymes complex I. Due to this defect, normal aerobic respiration is impaired which results in increased proliferation of mitochondria in the epithelial cells as a compensatory mechanism to supply ATP.
Oncocytomas are of epithelial origin and occur in thyroid, parathyroid, kidney, pituitary gland and salivary glands.
Parotid oncocytomas are associated with Birt-Hogg Dube syndrome (BHD). This syndrome was first described in 1977. The genetic alteration for BHD is mapped to chromosome 17p11.2. This gene codes for folliculin which is a tumour suppressor protein. This protein might also have a role in energy/nutrient sensing signalling pathways which is associated with ATP and mitochondria. The other features of this syndrome include benign proliferations of hair follicle and increased risk for kidney tumours [11,44].
Other Associated Syndromes
The oral manifestations of MEN 2B, HTP-JT, familial OSCC, neuro fibromatoses and the most common familial syndromes with oral stigmata are presented in [Table/Fig-3] [2,7,10,12,14].
Conclusion
Relatives of a person diagnosed with familial tumours should be examined for signs of the syndrome as he or she may be the first person to be affected. The mutations may be present in parents or siblings but they themselves may not be affected and may pass the condition on to their decedents. Depending on the syndrome, genetic professionals can determine who in the family is at risk. Many a times oral manifestations are the first feature, in conditions such as hyperparathyroidism associated ossifying fibroma (30%), leuloplakia with DC (80%), carotid body tumours, neurofibromas, Goltz-Gorlin and Gardner’s syndrome to name a few. Most often dentists are the first to encounter such manifestations and hence, play a major role in identifying the familial cancers.
[1]. NCI Dictionary of cancer terms [Internet]Last updated on 2015 April 10; Cited 2017 Feb 16], Last accessed on 2017 May 5Available from: http://www.cancer.gov/publications/dictionaries/cancer-terms?cdrid=256552 [Google Scholar]
[2]. Marsh DJ, Theodosopoulos G, Howell V, Richardson AL, Benn DE, Proos AL, Rapid mutation scanning of genes associated with familial cancer syndromes using denaturing high-performance liquid chromatographyNeoplasia 2001 3(3):236-44. [Google Scholar]
[3]. Longe JL, Gale Encyclopedia of Cancer: A guide to cancer and its treatments 2005 12nd edFarmington HillsThomson Gale:459 [Google Scholar]
[4]. Neel JV, Schull WJ, Human Heredity 1954 1st editionChicagoUniversity of Chicago Press [Google Scholar]
[5]. Kimbrough RD, Mehaffey KR, Grandjean P, Sandoe SH, Rustein DD, Clinical effects of environmental chemicalsA software approach to aetiological diagnosis 1989 1st edNew YorkHemisphere Publishing Corporation [Google Scholar]
[6]. Wikipedia: free enclyopediaLast updated on 2017 April 2; Last cited 2017 April 16, Last accessed on 2017 May 5Available from: https://en.wikipedia.org/wiki/Genomic_imprinting [Google Scholar]
[7]. Prime SS, Thakker NS, Pring M, Guest PG, Paterson IC, A review on inherited cancer syndromes and their relevance to oral squamous cell carcinomaOral Oncol 2001 37(1):01-16. [Google Scholar]
[8]. Schrader KA, Sharaf R, Alanee S, Offit K, Genetic Factors: Hereditary Cancer Predisposition Syndromes [Internet] 2015 updated 2015 March 4, cited 2016 November 27Available from: http://clinicalgate.com/genetic-factors-hereditary-cancer-predispositionsyndromes-16 [Google Scholar]
[9]. Duan K, Mete O, Familial hyperparathyroidism syndromesDiagnostic Histopathology 2016 22(3):92-100. [Google Scholar]
[10]. Suarez C, Rodrigo JP, Ferlito A, Cabanillas R, Shaha AR, Rinaldo A, Tumours of familial origin in the head and neckOral Oncol 2006 42:965-78. [Google Scholar]
[11]. Gasparre G, Romeo G, Rugolo M, Porcelli AM, Learning from oncocytic tumours: Why choose inefficient mitochondriaBiochimica et Biophysica Acta 2011 1807:633-42. [Google Scholar]
[12]. Ponti G, Tomas A, Manfredin M, Pellacani G, Oral mucosal stigmata in hereditary cancer syndromes: From germline mutations to distinctive clinical phenotypes and tailored therapiesGene 2016 582(1):23-32. [Google Scholar]
[13]. Perry A, Perry A, Brat DJ, Familial tumour syndromesPractical Surgical Neuropathology: A Diagnostic Approach 2010 1st edPiladelphiaChurchill Livingstone:427-453. [Google Scholar]
[14]. Pan E, Prados MD, Kufe DW, Pollock RE, Weichselbaum RR, Bast RC, Gansler TS, Holland JF, Familial Tumours Syndromes of the Central Nervous systemHolland-Frei Cancer Medicine 2003 6th edHamiltonBC Decker Inc [Google Scholar]
[15]. Thakker RV, Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4)Mol Cell Endocrinol 2014 386:02-15. [Google Scholar]
[16]. Spiegel AM, Focus on hereditary endocrine neoplasiaCancer Cell 2004 6:327-32. [Google Scholar]
[17]. MacIntosh RB, Shivapuja PK, Krzemien MB, Lee M, Multiple endocrine neoplasia type 2b: maxillofacial significance in 5 casesJ Oral Maxillofac Surg 2014 72(12):2498 [Google Scholar]
[18]. Winkelmann RK, Carney JA, Cutaneous neuropathology in multiple endocrine neoplasia, type 2bJ Invest Dermatol 1982 79:307-12. [Google Scholar]
[19]. Usami Y, Takenobu T, Kurihara R, Imai Y, Shinohara S, Fukuda Y, Neural hyperplasia in maxillary bone of multiple endocrine neoplasia type 2B patientOral Surg Oral Med Oral Pathol Oral Radiol Endod 2011 112:783-90. [Google Scholar]
[20]. Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT, Hyperparathyroidism-jaw tumour syndromeJournal of Internal Medicine 2003 253:634-42. [Google Scholar]
[21]. Yamashita Y, Akiyama T, Mizusawa N, Yoshimoto K, Goto M, A case of hyperparathyroidism jaw tumour syndrome found in the treatment of an ossifying fibroma in the maxillary bone. IntJ. Oral Maxillofac Surg 2007 36:365-69. [Google Scholar]
[22]. Gnepp DR, Diagnostic surgical pathology of head and neck 2009 2nd edPhiladelphiaSaunders, an imprint of Elsevier [Google Scholar]
[23]. Manvikar V, Kulkarni R, Koneru A, Vanishree M, Role of human papillomavirus and tumour suppressor genes in oral cancerJ Oral Maxillofac Pathol 2016 20:106-10. [Google Scholar]
[24]. Bertonhaa FB, Mateus de Camargo BF, Kuasnea H, Pintor dos Reisb P, Erika da Costa P, Munoza JJAM, PHF21B as a candidate tumour suppressor gene in head and neck squamous cell carcinomasMolecular Oncology 2015 9:450-62. [Google Scholar]
[25]. Noto Z, Tomihara K, Furukawa K, Noguchi M, Dyskeratosis congenital associated with leukoplakia of the tongueInt J Oral Maxillofac Surg 2016 45:760-63. [Google Scholar]
[26]. Bessler M, Wilson DB, Mason PJ, DyskeratosiscongenitaFEBS Lett 2010 584:3831-38. [Google Scholar]
[27]. Handley TPB, McCaul JA, Ogden GR, Dyskeratosis congenitalOral Oncol 2006 42:331-36. [Google Scholar]
[28]. Wei-Hua J, Bing-Jian F, Zong-Li X, Xiao-Shi Z, Huang P, Li-Xi H, Familial risk and clustering of nasopharyngeal carcinoma in Guangdong, ChinaCancer 2004 101:363-69. [Google Scholar]
[29]. Yang X, Scott Diehl S, Pfeiffer R, Chien-Jen C, Wan-Lun H, Dosemeci M, Evaluation of risk factors for nasopharyngeal carcinoma in high-risk nasopharyngeal carcinoma families in TaiwanCancer Epidemiol Biomarkers Prev 2005 14(4):900-05. [Google Scholar]
[30]. Loh KS, Goh BC, Lu J, Wen-Son H, Tan L, Familial nasopharyngeal carcinoma in a cohort of 200 patientsArch Otolaryngol Head Neck Surg 2006 132:82-85. [Google Scholar]
[31]. Soura E, Eliades PJ, Shannon K, Tsao H, Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndromeJ Am Acad Dermatol 2016 74(3):395-407. [Google Scholar]
[32]. Jenkins NC, Jung J, Liu T, Wilde M, Sheri L, Grossman H, Familial melanoma–associated mutations in p16 uncouple its tumour-suppressor functionsJ Invest Dermatol 2013 133:1043-51. [Google Scholar]
[33]. Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H, Hereditary melanoma: Update on syndromes and management: Emerging melanoma cancer complexes and genetic counsellingJ Am Acad Dermatol 2016 74(3):411-20. [Google Scholar]
[34]. Collins N, Dietzek A, Contiguous bilateral head and neck paragangliomas in a carrier of the SDHB germline mutationJ Vasc Surg 2012 55:216-19. [Google Scholar]
[35]. Mediouni A, Ammari S, Wassef M, Gimenez-Roqueploc AP, Laredo JD, Duet M, Malignant head/neck paragangliomas. Comparative StudyEur Ann Otorhinolaryngol Head Neck Dis 2014 131:159-66. [Google Scholar]
[36]. Shankavaram U, Fliedner SMJ, Elkahloun AG, Barb JJ, Munson PJ, Thanh T, Genotype and tumour locus determine expression profile of pseudohypoxic pheochromocytomas and paragangliomasNeoplasia 2013 15(4):435-47. [Google Scholar]
[37]. Javed F, Ramalingam S, Ahmed HB, Gupta B, Sundar C, Qadri T, Oral manifestations in patients with neurofibromatosis type-1: A comprehensive literature reviewCrit Rev Oncol Hematol 2014 91:123-29. [Google Scholar]
[38]. Rad E, Tee AR, Neurofibromatosis type 1: Fundamental insights into cell signalling and cancerSemin Cell Dev Biol 2016 52:39-46. [Google Scholar]
[39]. Martin AH, Rodriguez AD, An update on neurofibromatosis type I: not just cafe-aulait spots and freckling. Part II. Other skin manifestations characteristic of NF1.NF 1 and cancerActas Dermosifiliogr 2016 107(6):465-73. [Google Scholar]
[40]. Martin AH, Rodriguez AD, An update on neurofibromatosis type I: not just cafe-aulait spots and neurofibromas. Part I. Dermatological clinical criteria diagnostic of the diseasaeActas Dermosifiliogr 2016 107(6):454-64. [Google Scholar]
[41]. Tadini G, Milani D, Menni F, Pezzani L, Sabatini C, Esposito S, Is it time to change the neurofibromatosis 1 diagnostic criteria?Eur J Intern Med 2014 25:506-10. [Google Scholar]
[42]. Javed F, Ramalingamb S, Ahmed HB, Gupta B, Sundar C, Qadri T, Oral manifestations in patients with neurofibromatosis type-1: A comprehensive literature reviewCrit Rev Oncol Hematol 2014 91:123-29. [Google Scholar]
[43]. Radek M, Tomasik B, Wojdyn M, Snopkowska-Wiaderna D, Błaszczyk M, Radek A, Neurofibromatosis type 2 (NF 2) or schwannomatosis?–Case report study and diagnostic criteriaNeurologia I neurochirurgiapolska 2016 50:219-25. [Google Scholar]
[44]. Gasparre G, Romeo G, Rugolo M, Porcelli AM, Learning from oncocytic tumours: Why choose inefficient mitochondria?Biochimica et Biophysica Acta 2011 1807:633-42. [Google Scholar]