The clinical applications of antibacterial chemotherapy began in 1935 with the discovery of prontosil. Over the next three decades, virtually all the major classes of antibiotics and synthetic antibacterial agents in current use were introduced. The wide spread introduction of these antimicrobials into clinical practice have made possible the treatment of life threatening diseases and enabled advances in surgical techniques by permitting the prophylaxis and treatment of surgery related infections.
However, steadily increasing drug resistance in the treatment of infectious disease posed a serious problem in antimicrobial therapy and necessitates continuing research on different classes of thiadiazole derivatives. Thiadiazole compounds are known to act as antimicrobials [1,2], antituberculosis [3], anti-inflammatory [4,5], anticonvulsant [6,7], antihypertensive [8,9], local anesthetic and anticancer [10,11]. The 1,3,4-thiadiazoles are known to exhibit various biological activities due to the presence of N=C-S moiety [12].
Moreover, numerous therapeutically important medicines like terconazole, itraconazole, fluconazole, cefazoline are known to contain one of these heterocyclic nucleus. In view of the above mentioned facts and in continuation of our work on the synthesis of the effective treatment of human diseases, the synthesis of some novel 2, 5-disubstituted-1,3,4-thiadiazole derivatives was carried out. The reaction sequence leading to the formation of desired heterocyclic compounds are outlined in [Table/Fig-1].
Generally, compounds subsequent to synthesis are subject to extensive proteomic approach to identify potential binding proteins. Such a technique involves comparison of the protein expression profiles for a given cell or tissue in the presence or absence of the given molecule. This method has not proved very successful in target discovery because it is laborious and time consuming [13,14]. Alternatively, we have implemented in silico target profiling method in identification of potential targets using online server PharmMapper [13]. Virtual screening method was further applied to predict binding free energies of synthesized compounds using AutoDock 4.2. Extensive post virtual screening analysis of docked complexes was carried out on basis of MHP to evaluate hydrophobic contribution of synthesized compounds to ligand binding site of identified target protein.
Materials and Methods
The chemical synthesis of the di substituted thiadiazole compounds was conducted at Department of Chemistry, Padmashree Vikhe Patil College, Loni, Maharashtra, India during the month of September 2015-December 2015 and computational analysis was carried out at the Bioinformatics Centre, Savitribai Phule Pune University, Pune, Maharashtra, India during month of May 2016-June 2016. The present data is generated at respective laboratories with permission of the departmental heads. Since no animal models were involved in the present study, hence there was no need of ethical approval.
Chemical Synthesis
Melting points were taken in open capillaries and were uncorrected. Purity of the compounds was checked on TLC. IR spectra were recorded on a Perkin Elmer 1420 FT spectrophotometer in KBr disc [1]. H NMR spectra were recorded on a Bruker avance II 400 MHz spectrometer using CDCl3 as a solvent and TMS as internal standard. Peak values are shown in δ ppm. Mass spectra were recorded on a Waters mass spectrometer.
5-((2,5-dichlorophenoxy)methyl)-N-phenyl-1,3, 4-thiadiazol-2-amine (4a-i)
Thiosemicarbazide derivative 3 (0.001 mol) was dissolved in concentrated H2SO4 (10 ml) in 50 ml round bottom flask and stirred at room temperature for three hours. After completion of reaction 15 g of crushed ice was added. The solid obtained was separated by filtration and crystallized from 1:1 mixture of DMF and water to afford thiadiazoles 4. The compounds synthesized by above method are listed in [Table/Fig-2].
Data of synthesized thiadiazole compounds.
| Sl. No | R1 | R2 | R3 | R4 | M.PC | Traditional | Ultra-sonication |
|---|
| Time(hr) | Yield (%) | Time(min) | Yield (%) |
|---|
| 4a | H | H | H | H | 278 | 3 | 54 | 30 | 63 |
| 4b | Cl | Cl | H | H | 184 | 3 | 52 | 28 | 60 |
| 4c | Cl | H | Cl | H | 256 | 3 | 55 | 27 | 61 |
| 4d | H | Cl | H | Cl | 238 | 3 | 53 | 29 | 62 |
| 4e | H | OCH3 | H | H | 212 | 3 | 52 | 28 | 60 |
| 4f | H | H | CH3 | H | 160 | 3 | 53 | 28 | 63 |
| 4g | CH3 | H | H | H | 206 | 3 | 54 | 28 | 65 |
| 4h | H | H | Cl | H | 210 | 3 | 53 | 30 | 64 |
| 4i | H | H | F | H | 252 | 3 | 55 | 30 | 65 |
By ultrasound method: Thiosemicarbazide derivative 3 (0.001 mol) and conc. H2SO4 (10 ml) were taken in 100 ml RBF. The reaction mixture was subjected to ultrasound irradiation for 25-30 min. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was poured into crushed ice. The product obtained was separated by filtration and crystallized from DMF/water to afford the compound 4. The compounds synthesized by above procedure are listed in [Table/Fig-2] and their structures are provided in [Table/Fig-3].
Chemical structures of nine novel 2, 5-di substituted-1,3,4-thiadiazole derivatives synthesized in this study.
4f: IR (KBr) cm-1 : 3266 (NH), 1560(C=N), 1189 (Ar-Cl); 1H NMR (CDCl3): 2.25 (s, 3H, Ar-CH3), 7.10-8.00 (m, 7H, Ar-H), 9.75 (s,1H, NH); Mass: (M+); m/z 366 .
Pharmacophore Analysis
Pharmacophore matching approach was utilized to identify targets for novel synthetic derivatives using PharmMapper server [13]. Initially, compounds were obtained in sdf format to upload on PharmMapper server. Multiple conformers, but not more than 300, were allowed to be generated. Only human protein target set was utilized for pharmacophore mapping in this study. Rest of parameters was set to default values. Further detailed pharmacophore analysis and alignment were performed using LigandScout 2.0 package [15].
Preparation of Compounds for Molecular Modeling
The 2D structures of above synthesized thiadiazole compounds were drawn in ChemDraw® 8.0 (CambridgeSoft, Cambridge, MA, USA) and their SMILES were obtained. Following to this step, 3D conformers of these compounds were generated in sdf format using FROG2 server and AutoDock4.2 available from Python Prescription 0.8 (PyRx) was used for molecular docking analysis [16,17]. Open Babel utility in PyRx environment was used to import ligand molecules in sdf format for subsequent energy minimization using UFF force field [18-20]. All these molecules were minimized for over 200 steps using conjugate gradient optimization algorithm. Molecules were updated at every step during the energy minimization.
Testing Validity of AutoDock 4.2 and Virtual Screening
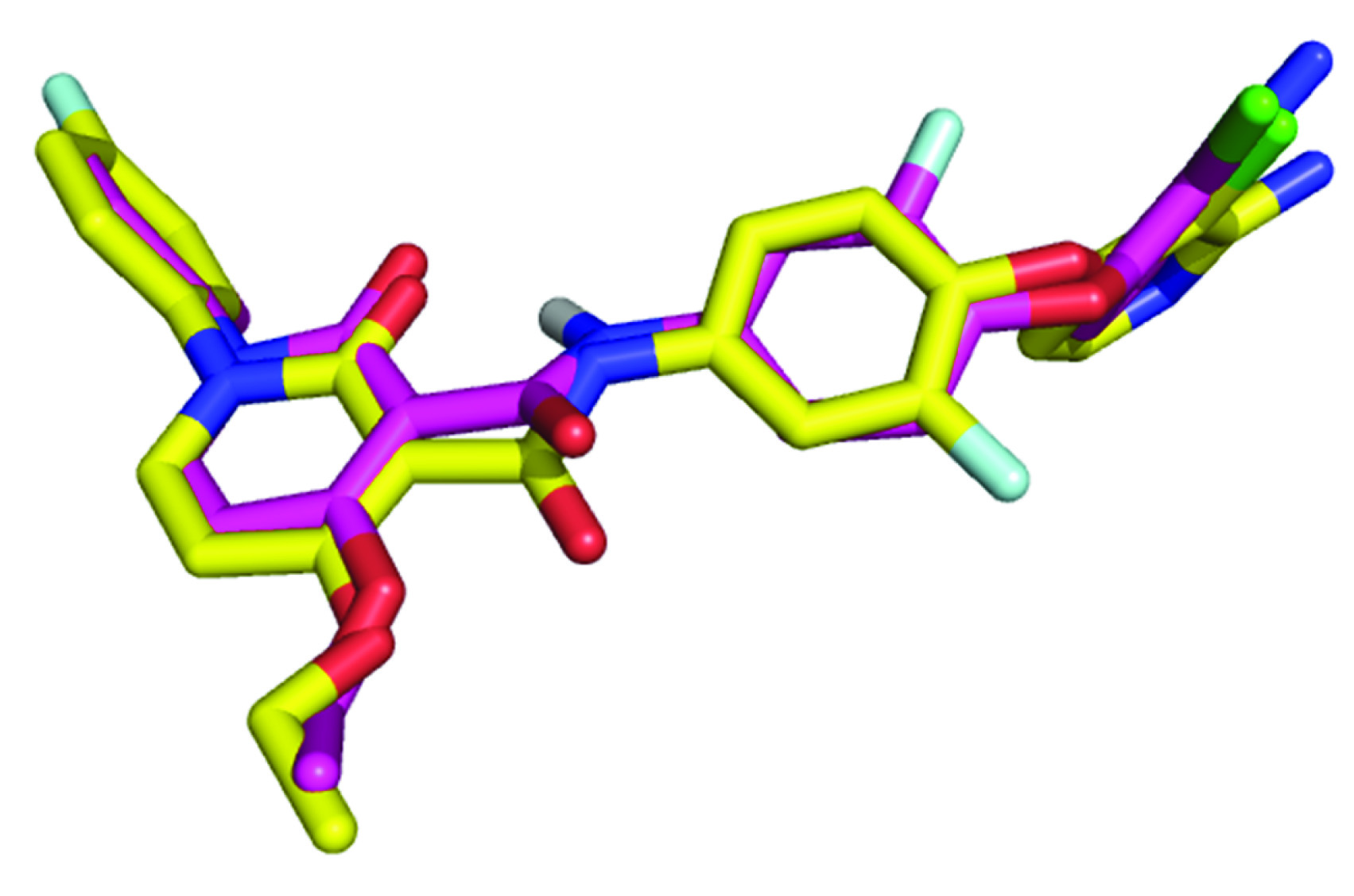
The validity of a docking system can be checked by testing the ability of a docking algorithm to reproduce the experimental binding mode of a ligand. After docking, Root Mean Square Deviation (RMSD) value of the predicted pose to experimentally verified pose is calculated. The obtained RMSD values are well under 2 Å that clearly indicates successful prediction of binding [21]. The grid files were obtained using Auto-grid program and the affinity grid of 50 × 50 × 50 points was set using spacing of 0.375 Å to cover entire active site. The conformational search for obtaining optimal binding pose was carried out using the lamarckian genetic algorithm. Each lamarckian job was set to have 10 runs and restricting the initial population to 150 structures. The maximum number of energy evaluation and generation were set to 27000. Single top individual was allowed to survive to next generation, rate of gene mutation and crossover was set to 0.02 and 0.8 respectively and the rest of parameters were set to default values. The final structures were clustered according to native autodock scoring function. The top ranked conformations of each ligand were selected. RMSD value of 0.87 Å was obtained from the docking experiment of crystallographic ligand BMS-777607 analog back in ligand binding site of human hepatocyte growth factor receptor. This value indicates that predicted binding mode is nearly identical to the X-Ray crystallography conformer [Table/Fig-4]. Same set of parameters were used for virtual screening of above synthesized compounds.
Comparison of re-docking results of ligand to X-Ray crystallographic mode of binding (model with magenta colored carbons in sticks represent docking result while model colored in yellow is experimentally verified binding pose).
Post Virtual Screening Analysis
Best scoring docking poses were further evaluated on basis of hydrophobic interaction employing an online server Protein Ligand Atractions Investigation Numerically (PLATINUM) [22] by calculating Molecular Hydrophobic Potentials (MHP). PyMol was utilized to visualize the resulting structures and MHP data. Detailed interactions of thiadiazole compounds with c-Met receptor were inferred from a JAVA based GUI of LigPlot program called LigPlot+ [23,24].
Results
Target Identification
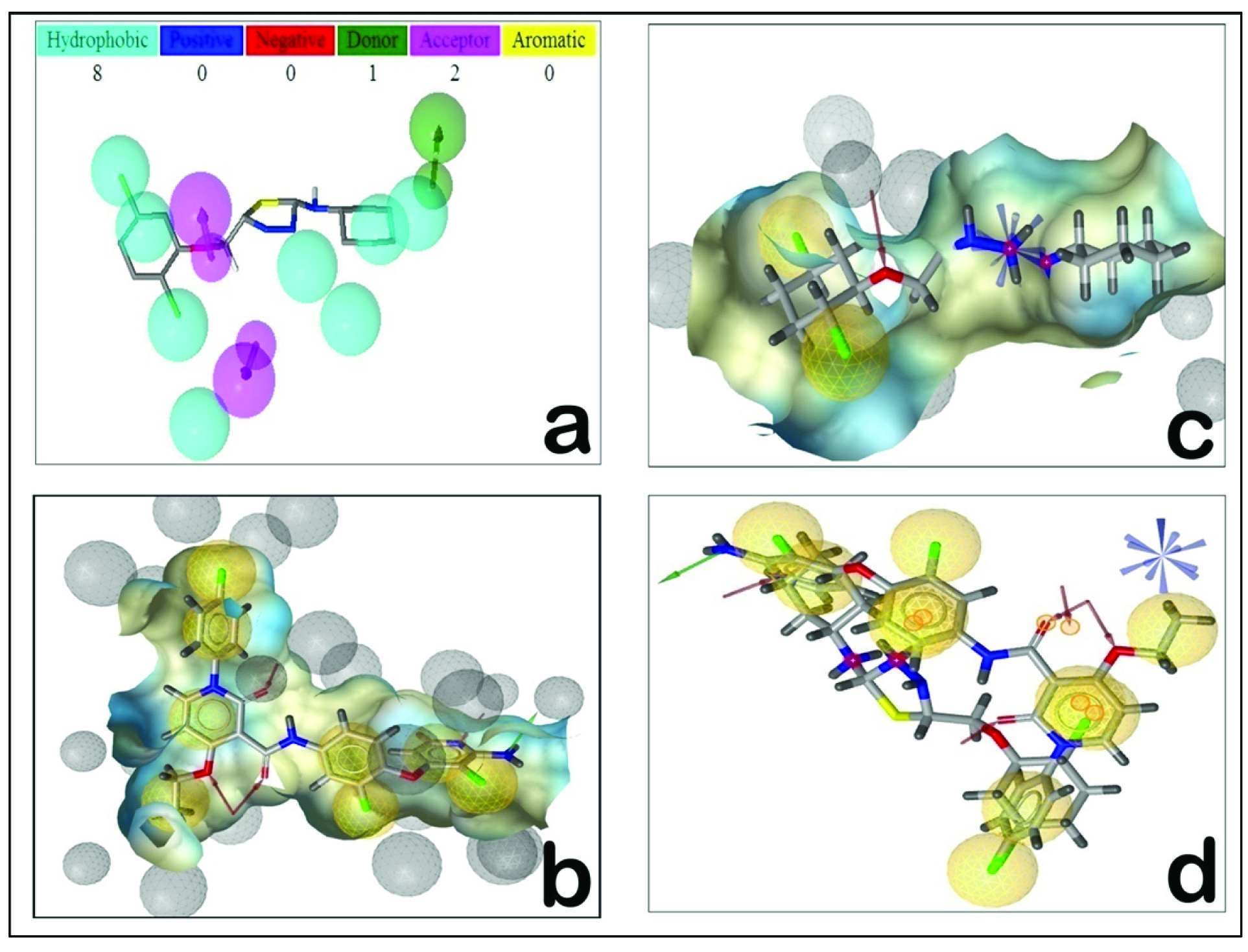
Pharmacophore is the 3D orientation of the functional groups of a molecule that interacts with target protein [13]. PharmMapper server works by ‘probing’ the ligand into a database of pharmacophore models of binding sites. It functions on the ligand-protein reverse docking strategy and reports potential target on the basis of normalized fit score [25,26]. In addition to that, we have calculated z’-score for better statistical meaning and confidence while comparing obtained results. Large positive z’-score of identified target indicates high significance of the target to a query compound, on contrary, large negative z’-score indicates that the target identified is not significant. [Table/Fig-5] describes top ranked targets obtained from server along with their normalized fit score and z’-score. Pharmmapper identified hepatocyte growth factor receptor (c-Met) among top three candidates as potential targets for thiadiazole derivatives with fit score of 4.306 and z’-score of 1.52. Comparison of normalized fit score and z’-score obtained from pharmacophore mapping analysis indicate that human c-Met receptor with PDB ID 3F82 might act as potential target. Pharmmapper detected eleven featured pharmacophore model for thiadiazole derivatives that consist of eight hydrophobic areas around terminal dichlorophenyl and phenyl moieties [Table/Fig-6a]. Two hydrogen bond acceptor features are detected around methoxy region, while a single hydrogen bond donor feature was observed around amine of centrally placed thiadiazole moiety. This pharmacophore model is consistent with experimental observation that identified c-Met receptor favours hydrophobic ligands, since its ligand binding site is occupied by majority of lipophilic residues [27]. Pharmacophores can be aligned to gain insights of more therapeutically significant features among multiple pharmacophore models [28]. Pharmacophore alignments thus generate an efficient and consensus set of features that identify the specific characters that are required for a ligand to interact with a precise target receptor [29-35]. Detail pharmacophore analysis was carried out by aligning the pharmacophores using Ligandscout 2.0 package. In order to align the predicted pharmacophore from pharmmaper server, the pharmacophore model was first constructed for the experimentally verified inhibitor of c-Met receptor. Pharacophore model generated for experimentally crystallized ligand from PDBID 3F82 consist of 13 features including eight hydrophobic contacts, four hydrogen bond acceptors and a single hydrogen bond donor feature [Table/Fig-6b]. Pharmacophore model of representative thiadiazole compound and further alignment results clarify the facts that two hydrophobic features along with single HBA feature might be essential chemical requirements for recognition as c-Met inhibitor [Table/Fig-6c,d]. Additionally this pharmacophore model can prove to be a good starting point for optimizing further chemical synthesis of compound aimed to inhibit c-Met receptor using thiadiazole as parent scaffold.
Top target hits obtained by pharmacophore mapping using Pharm Mapper server.
| Target Name | PDB ID | Number of Pharmacophore Features | z’-score |
|---|
| Hepatocyte growth factor receptor | 3F82 | 11 | 1.52a |
| Cell division protein kinase 2 | 1V1K | 6 | 1.47 |
| VEGF receptor 2 | 2OH4 | 8 | 1.44 |
| Renin | 2V12 | 9 | 1.35 |
| Beta secretase I | 2QK5 | 7 | 1.32 |
a Target showing comparatively best z’-score.
Figure representing pharmacophore data: Pharmacophore model obtained from PDB ID 3F82 and thiadiazole (b and c respectively) and pharmacophore alignment (d). Note the aligned pharmacophore features are highlighted in small yellow circles (Fig d). Fig a was obtained from pharmmapper, while Fig b, c and d were obtained using LigandScout 2.0.
Structural Analysis of Docked complexes
To further cross validate the results obtained from PharmMapper and Ligand Scout regarding suitability of synthesized thiadiazole compounds for c-Met receptor, we further performed molecular docking investigation. Moreover, structural analysis of docking complexes provides valuable information regarding possible interactions that given compounds might form at binding site of c-Met receptor. This information thus confirms suitability of given compounds for identified receptor. Results obtained from docking experiment observe that compounds 4c, 4e and 4h are found to be in van der Waals contact with residue Ile-1084 [Table/Fig-7]. In crystallographic analysis reported by Schiering N et al., Ile-1084 is known to hydrophobically interact with inhibitor K-252a with a high complementarity between the interacting surfaces [36]. Similarly the tri-cyclic ring of inhibitor MK-2461 analog is characterized to forms a hydrophobic contact with Ile-1084 [37]. These facts point out that interaction of compounds 4c, 4e and 4h with Ile-1084 might act as crucial factor for c-Met inhibition. All the compounds except 4d and 4h are observed to make liphophilic interaction with residue Ala-1108 [Table/Fig-7]. This interaction is experimentally verified to be vital in inhibition of c-Met receptor by inhibitors like K-252a and MK-2461 analog [36,37] pointing towards the possibility of interaction of these compounds with Ala-1108.
Docking results for synthesized thiadiazole derivatives in human c-Met receptor, by AutoDock 4.2.
| Ligand name | Binding Free Energy (Kcal/mol) | Interacting Amino acids (HBD Å) | No. of HB (Average distance) | HD::HA | Residues in hydrophobic/ van der Waals interaction |
|---|
| 4a | -8.38 | -- | -- | -- | Ala1108, Met1131, Phe1134, Leu1140, Leu1157, Pro1158, Met1160, Met1211, Ala1221, Asp1222, Phe1223 |
| 4b | -8.84 | Asp1222 | 1 (3.08) | Asp1222 (Nbb):H :: Lig(O):O1 | Ala1108, Lys1110, Glu1127, Leu1140, Leu1157, Pro1158, Met1160, Met1211, Ala1221, Phe1223, Met1131, |
| 4c | -8.28 | Asp1222 | 1 (3.20) | Asp1222 (Nbb):H :: Lig(O):O1 | Ile1084, Lys1110, Glu1127, Met1131, Leu1140, Leu1157, Ala1221, Met1211, Ala1108, Phe1223 |
| 4d | -8.18 | Asp1222 | 2 (2.54) | Lig(N1):H :: Asp1222 (Obb):OLig(N2):H :: Asp1222 (Obb):O | Lys1110, Met1131, Leu1140, Val1155, Leu1157, Phe1200, Met1211, Ala1221 |
| 4e | -8.43 | -- | -- | -- | Ile1084, Ala1108, Met1131, Phe1134, Leu1140, Leu1157, Pro1158, Tyr1159, Met1160, Met1211, Asp1222, Phe1223 |
| 4f | -8.29 | -- | -- | -- | Ala1108, Lys1110, Met1131, Phe1134, Leu1140, Leu1157, Pro1158, Met1160, Phe1200, Ala1221, Asp1222, Phe1223 |
| 4g | -8.39 | Asp1222 | 1 (3.20) | Asp1222 (Nbb):H :: Lig(S):S1 | Val1092, Ala1108, Met1131, Phe1134, Val1139, Leu1140, Leu1157, Phe1200, His1202, Val1220, Phe1223, |
| 4h | -8.51 | -- | -- | -- | Ile1084, Val1092, Lys1110, Ala1221, Met1131, Phe1134, Leu1140, Leu1157, Phe1200, Asp1222, Phe1223 |
| 4i | -8.02 | -- | 2 (2.94) | Asp1222 (Nbb):H :: Lig(O):O1Met1160(Nbb) H :: Lig(F):F1 | Ala1108, Val1092, Met1131, Phe1134, Leu1140, Leu1157, Pro1158, Met1211, Ala1221, Phe1223 |
Discussion
Compounds 4g, 4h and 4i are found to make van der Waals contact with residue Val-1092 [Table/Fig-7]. In an experimental inhibition study of c-Met by inhibitor AM7, Val-1092 was located almost directly above the inhibitor AM7 and forms van der Waals contacts with it and hence suspected to play a key role in binding of inhibitor [38], additionally hydrophobic contribution of Val-1092 is experimentally verified to play an important role in inhibition by compound K-252a [36]. These points suggest possibility of involvement of Val-1092 in inhibition of c-Met receptor by compounds 4g, 4h and 4i.
Lys-1110 is found to be interacting with compound 4b, 4c, 4d, 4f and 4h. Lys-1110 is known to play a role in stabilizing the particular conformation of the activation loop. Conserved residue Lys-1110 forms hydrogen bond interaction with Asp-1228 and keeps the activation loop in a confirmation that facilitate the vital pi- stacking interaction between residue Tyr-1230 and inhibitor NVP-BVU972 in process of inhibition [39]. This residue is characterized to hydrophobically interact with inhibitor K-252a and MK-2461 [37]; additionally the salt bridge between residues Lys-1110 and Glu-1127 is already known to maintain active kinase conformation [36]. Upon binding of inhibitor ARQ 197 to c-Met receptor, the alkyl side chain of Lys1110 get twisted and participates in a hydrophobic interaction with the tri-cyclic moiety of ARQ 197 [40]. All these observations indicate the possibility that compounds 4b, 4c, 4d, 4f and 4h might interact with Lys-1110 if used in c-Met inhibition.
Compound 4b and 4c are found to be interacting with Glu-1127 [Table/Fig-7]. Glu-1127 dynamically determines kinase activity of c-Met receptor by maintaining its phosphorylated or unphosphorylated conditions [37]. The autophosphorylation event in activation loop triggers the kinase activity of c-Met. The activation loop is placed against the active site in the unphosphorylated state of enzyme, thus interrupting the catalytically capable conformation of the kinase domain; ultimately congesting the ATP binding site [41]. The act of autophosphorylation alleviates the auto-inhibitory characters of c-Met by eviction and dislocating the activation loop [37]. Principally, when the G loop takes up the extended conformation along with helix C and the catalytic loop reassembles, the ATP binding site is unveiled to form a viable active site. In the unphosphorylated form of c-Met, Tyr-1235 remains buried forming a hydrogen bond with Glu-1127 [41]. Inhibitory analog of compound MK-2461 imparts conformational change in the G loop leading to formation of a salt bridge between Glu-1127 and conserved Lys-1110 instead of hydrogen bonding with Tyr-1235 [37]. This particular interaction with Glu-1127 is experimentally proved to be important in catalysis as observed from differential binding study of AM7 [38]. Binding of inhibitor K-252a is known to induce an inhibitor confirmation in the receptor, disorienting the catalytic positioning of Glu-1127 ultimately inhibiting nucleotide binding [36]. In c-Met inhibition study involving inhibitor EXEL-2880, Glu-1127 plays a pivotal role by forming a hydrogen bond with the inhibitor [42]. Eathiraj S et al., suggested that in addition to phosphorylation- dephosphorylation events, C-helix is actively involved in regulation of kinase activity of c-Met [40]. The C-helix is located in very close vicinity of ATP binding pocket and thus, plays a key role in sustaining the ion-pair interaction among the functionally conserved Glu-1127 and Lys-1110 residues. Therapeutic agent ARQ 197 is known to stabilize the inactive conformation of c-Met by breaking the ion-pair interaction between Glu-1127 and Lys-1110 at the ATP-binding cleft involving inhibitor induced structural rearrangement [40]. Thus, like inhibitors discussed above (MK-2461 analog, AM7, K-252a, EXEL-2880 and ARQ 197), compounds 4b and 4c by interacting with Glu-1127 might act as an efficient c-Met receptor inhibitor.
All of the synthesized thiadiazole compounds except 4b are observed to actively interact with residue Met-1131 [Table/Fig-7]. Schiering N et al., suspected that M1131T point mutation result in kinase activation via destabilization of inactive confirmation of c-Met, a leading feature of neoplasia [36]. In the inactive state of c-Met, Met-1131 is directly engage in the hydrophobic pocket accommodating the Phe-1223 side chain and simultaneously forms a van der Waals contact with Leu-1225. Their further site directed mutagenesis experiment of M1131T mutation confirmed that replacement of Met-1131 by threonine abate the hydrophobic contact making inactive confirmation thermodynamically less favourable, conclusively translating c-Met to an active kinase conformation [36]. Another evidence of critical importance of hydrophobic interaction of Met-1131 was clearly demonstrated in inhibition study of c-Met via compound LY2801653 [43]. In the Met-1131 mutant used in this study, due to change in hydrophobic side chain to polar and coupled size reduction resulted in weakened inhibitor binding [43]. The observation that majority of thiadiazole compounds displays hydrophobic contact with M-1131 [Table/Fig-7] is inductive of fact that these compounds may perform lipophily mediated inhibition of c-Met.
All the thiadiazole derivatives synthesized in this study appear to make hydrophobic interface with Leu-1140 [Table/Fig-7]. Such an interaction with Leu-1140 is published to play a critical role in inhibition of c-Met using compounds like K-252a [36] and MK-2461 [37]. Compound 4d is found to selectively interact with Val-1155. Functionally, Val-1155 and Phe-1200 actively form lipophilic cluster along with activation loop residues Leu-1225 and Phe-1223. This hydrophobic interaction occurs to be of imperative significance for maintaining specific position of the activation loop for crucial Pi-stacking interaction amidst inhibitor NVP-BVU972 and Tyr-1230 [39]. V1155L mutations are frequently observed in AMG458 resistance and also implicated in NVP-BVU972 and PF-02341066 resistance [39]. Moreover, Val-1155, an integral part of C-helix pocket is characterized to play a key structural role in binding and inhibition of drug-like lead compound AM7 [38].
All the selected thiadiazole compounds seem to be present in hydrophobic contact with residue Leu-1157 located in hinge region of c-Met receptor [Table/Fig-7]. Crystallographic analysis involving inhibition of c-Met via compound K-252a demonstrated influential role of such contact in stabilization and inhibition [36]. Thiadiazole compounds 4a, 4b, 4e, 4f and 4i are observed to be in close association with Pro-1158 via lipophilic contact [Table/Fig-7]. Pro-1158 plays critical role in inhibition of c-Met by inhibitor ARQ-197. Pro-1158 is known to make hydrogen bonds via its carbonyl oxygen of the main chain with characteristic canonical H-bond hinge interactions within indole ring of inhibitor. In another study involving inhibition of c-Met by an alkaloid compound K-252a, Pro-1158 was characterized to play a vital role in stabilization of enzyme-inhibitor complex by donating hydrogen bond to the lactam amide nitrogen of the inhibitor [40]. Thus, it is evident that residues like Leu-1140, Val-1155, Leu-1157 and Pro-1158 are most likely to participate in c-Met inhibition by synthesized thiadiazole compounds.
Thiadiazole compound 4e is uniquely found to be in hydrophobic contact with residue Tyr-1159 [Table/Fig-7]. Tyr-1159 holds a very key therapeutic position since point mutation at this location is implicated in resistance to drug AMG458 as it forms a direct interaction with quinoline ring of inhibitor AMG458 [39]. Tyr-1159 from hinge region is known to form apolar contact with inhibitor K-252a [36]. Derivative 4a, 4b, 4e and 4f seems to be actively associated to residue Met-1160 with hydrophobic contact, while compound 4i is observed to participate in polar interaction forming a hydrogen bond with backbone amide nitrogen of Met-1160 by means of its fluorine atom [Table/Fig-7,8]. This residue is experimentally characterized to form a hydrogen bond via its backbone amide with the lactum carbonyl oxygen of inhibitor K-252a [36], pyrrolidine-2,5-dione ring system of ARQ-197 [40] and EXEL-2880 [42]. Additionally Met-1160 is also known to make hydrophobic contact with tricyclic ring of analog of inhibitor MK-2461 [37]. Synthesized compounds 4d, 4f, 4g and 4h are noticed to be in hydrophobic contact with residue Phe-1200 [Table/Fig-7]. Phe-1200 is functionally analogous to Val-1155 discussed previously. F1200I mutation is involved in number of drug resistance mechanisms, mainly NVP-BVU972 and AMG 458 resistance [39]. In a c-Met inhibition study, Phe-1200 is recognized to directly interact with dihydropyrazolone moiety of AMG 458 [39]. F1200I mutation was found to be associated with enhanced phosphorylation of c-Met [39]. Western blot analysis carried out in same study indicates that F1200I mutation has tendency to interfere with the potency of both compounds (NVP-BVU972 and AMG458) to inhibit phosphorylation [39]. Kinetic assays evince that F1200I mutation lowers the Km values by six folds for ATP as compared to wild-type c-Met [39]. All these points clearly imply the possibility of involvement of residues like Tyr-1159, Met-1160, and Phe-1200 in thiadiazole based c-Met inhibition.
Figure showing docked complex of c-Met receptor (orange cartoons) with thiadiazole compounds (yellow sticks) making hydrogen bonds (magenta lines) with residues at the binding site (green sticks)
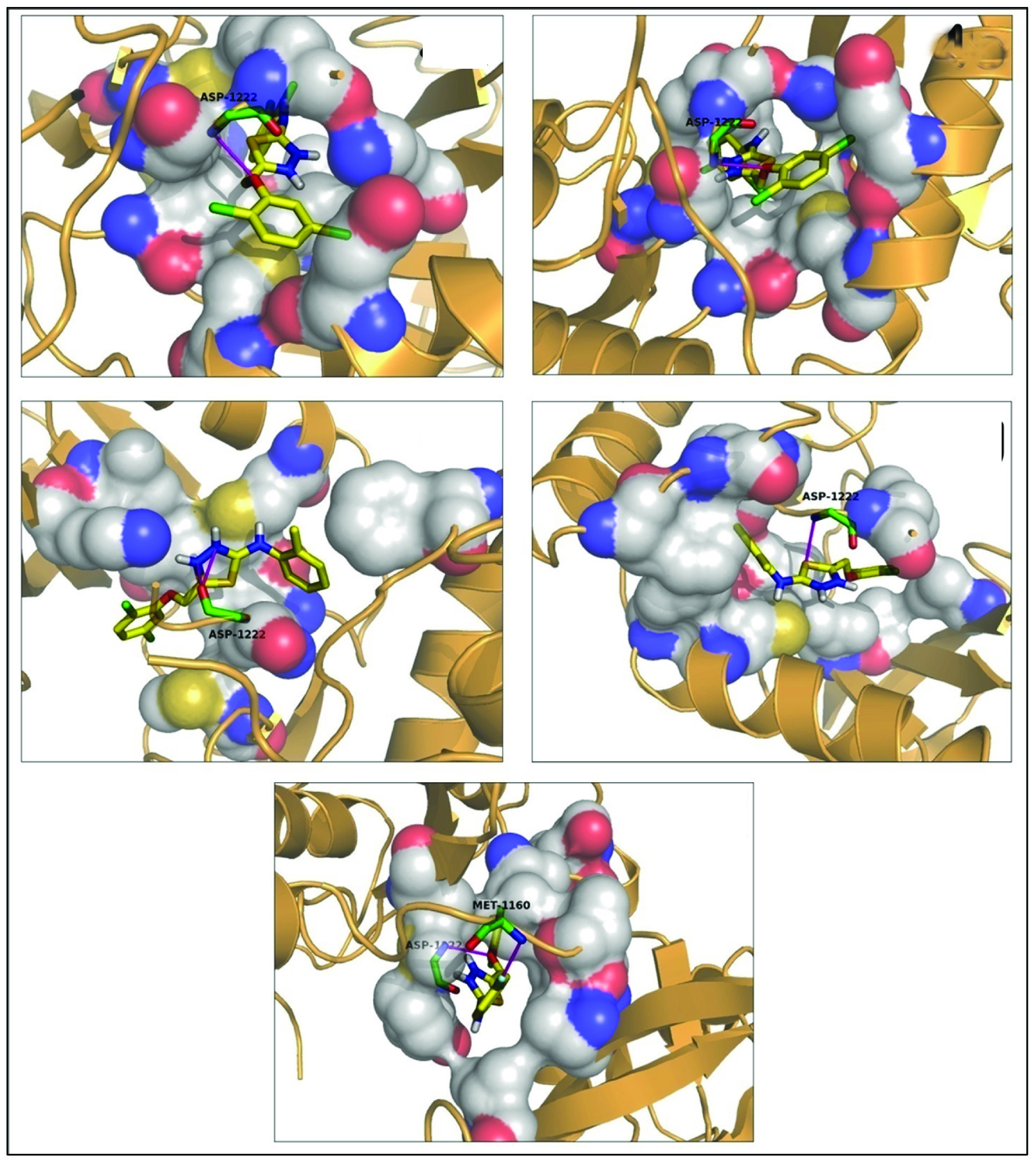
Derivatives generated from thiadiazole scaffold like compound 4a, 4b, 4c, 4d and 4i are found to be in van der Waals distance of Met-1211[Table/Fig-7]. Met-1211 is characterized to play pivotal role in inhibition of c-Met via inhibitors like SU11274 [38], AMG 458 [39] and MK-2461 [37]. Side chain of Met-1211 is detected to be involved in forming a platform for bottom of the indolocarbazole plane of inhibitor K-252a in the binding site of c-Met receptor [36]. Compound 4g exclusively interacts with residue Val-1220 of c-Met making a hydrophobic contact [Table/Fig-7]. Val-1220 is functionally involved in a hydrophobic interface between the N-lobe, C-lobe, and A-loop of c-Met receptor. Mutation at this position is believed to govern the conformational adaptations and transition event from inactive to an active kinase conformation of receptor [36]. Val-1220 is actively involved in inhibition by drug LY2801653. V1220I mutagenesis experiment showed noticeable decreases in IC50 values. This mutation is thought to slightly increase the bulk of the hydrophobic side chain, likely leading to stronger Van der Waals and/or hydrophobic interactions with the inhibitor [43]. Residue Ala-1221 is perceived to be in hydrophobic contact with majority of thiadiazole compounds (except 4e and 4g, see [Table/Fig-7]). Hydrophobic contribution of this residue was observed to play a decisive role in inhibition of c-Met by inhibitor MK-2461 [37]. Therefore, it can be convincingly predicted that residues like Met-1211, Val-1220, and Ala-1221 might involve in inhibitory activity of c-Met via present thiadiazole compounds.
Molecular docking results detected that all of the synthesized thiadiazole derivatives forms contact (either polar or hydrophobic) with conserved residue Asp-1222 [Table/Fig-7,8]. Asp-1222 is placed at first position in the activation loop and hence holds a very significant functional role not only in catalysis but also in inhibition process. Differential binding study of inhibitor AM7 proved that binding of AM7 displaces Asp-1222 away from active site and avoids its coordination with Mg ion ultimately inactivating it; demonstrating its influential role in catalysis [38]. In another c-Met inhibitory analysis by inhibitor K-252a cleared the fact that binding of inhibitor K-252a induces conformational changes that ultimately result in inhibition of nucleotide binding activity [36]. Majority of synthesized thiadiazole derivatives (except 4d) are found to be in hydrophobic contact with Phe-1223 [Table/Fig-7]. Phe-1223 governs numerous functional aspects of c-Met, for example, hydrophobic contribution of this residue is important in maintaining inactive kinase confirmation of c-Met demonstrating key role in cancer progression [36]. Displacement of Phe-1223 is essential for binding of inhibitors like AM7 to DEG pocket for inhibition [38]. Phe-1223 is known to hydrophobically interact with Val-1155 and Phe-1200. This hydrophobic contact facilitates the essential pi-stacking interaction between Tyr-1230 and NVP-BVU972 inhibitor [39]. In case of inhibition by ARQ 197, the c-Met-ARQ 197 complex adopts the DFG-out conformation where Phe-1223 sandwiches the tri-cyclic moiety of ARQ 197 and stabilizes the inactive conformation [40]. Inhibitor EXEL-2880 is known to dislodge Phe-1223 from its binding pocket in the apo-conformation and causes reorganization of the activation loop, such that Phe-1223 stacks with the fluorophenyl ring of the inhibitor. This is essential feature of the famous “phe in - phe out” mechanism of inhibition [42]. Synthesized compound 4b distinctively forms hydrophobic contact with Met-1131. M1131T point mutation is suspected to result in kinase activation, a hallmark of cancer. Mutation in Met-1131 is suspected to destabilize inactive c-Met confirmation. Met-1131 is known to directly participate in the hydrophobic pocket harboring the Phe-1223 side chain and is in van der Waals contact with Leu-1225. Replacing these residues by threonine diminished the hydrophobic contacts and making inactive conformation less favourable. Hence, mutation in this residue aids in the transition to an active kinase conformation [36]. Additionally, we also observed some novel interactions involving residues like Phe-1134, Val-1139, His-1202 never reported earlier.
Molecular Hydrophobic Potential Analysis
As observed from molecular docking results and above cited literature, hydrophobicity between ligand and receptor seems to play a very important role in inhibition of c-Met receptor. For example, in case of thiadiazole derivatives, compound showing one of the best docking score (binding free energy value of -8.51 Kcal/mol for compound 4h) is dominantly stabilized by hydrophobic interaction. Moreover, only five out of eleven thiadiazole compounds appear to make polar interaction (in form of hydrogen bonds) with c-Met receptor. Remaining six compounds are solely stabilized by hydrophobic forces. Therefore it becomes of paramount interest to evaluate hydrophobic contribution of thiadiazoles to c-Met receptor. Hydrophobic effect is successfully applied in various modeling studies since it is known to have an imperative role in recognition of inhibitor to its target receptor and provide strong backing for 3D structure of complex molecular assemblies [44,45]. Description of hydrophobic effect in macromolecules like proteins is popularly calculated by an empirical method called MHP [46]. Algorithm designed on this concept is implemented in a web based tool PLATINUM used to analyse and visualize hydrophobic/hydrophilic properties of biomolecules [22]. PLATINUM appraises the hydrophobic/hydrophilic interdependency of a ligand to the receptor-binding region; and hence, effectively determines hydrophobic/hydrophilic properties of both the ligand and its environment on the ligand molecular surface (the interface) [47]. According to MHP ideology, two hydrophobic or hydrophilic receptor-ligand contacts are considered as favourable or complementary, while hydrophobic-hydrophilic contacts are termed as unfavourable (or anticomplementary). Based on these basic assumptions, terms describing relative hydrophobicity of ligand to receptor are calculated that includes lipophilic match surface (SLL), hydrophilic match surface (SHH), buried surface area (Sburied), total surface area (Stotal). Another direct measure that indicates relative hydrophobicity of ligand to receptor is calculated as ‘fraction of matching hydrophobic surface’. This term is scored in range of zero to one, where score of one indicate total preeminence of hydrophobicity while score of zero implies complete hydrophilicity. MHP data for promising thiadiazole derivatives is tabulated in [Table/Fig-9]. Values from this table clearly demonstrate exemplary hydrophobic complementarity of thiadiazole ligands towards c-Met binding site.
MHP results of thiadiazole derivatives with human c-Met receptor using PLATINUM Server.
| Compound name | Area oflipophilicMatch(SLL) | Area ofhydrophilicmatch(SHH) | Buriedsurface area (Sburied) | Totalsurface area(Stotal) | Fraction ofmatchinghydrophobicsurface |
|---|
| 4b | 129.49 | 0.76 | 252.97 | 273.35 | 0.6441 |
| 4e | 145.19 | 1.58 | 282.40 | 302.41 | 0.6510 |
| 4g | 159.55 | 1.96 | 272.72 | 291.37 | 0.7108 |
| 4h | 166.33 | 0.44 | 254.49 | 280.25 | 0.7456 |
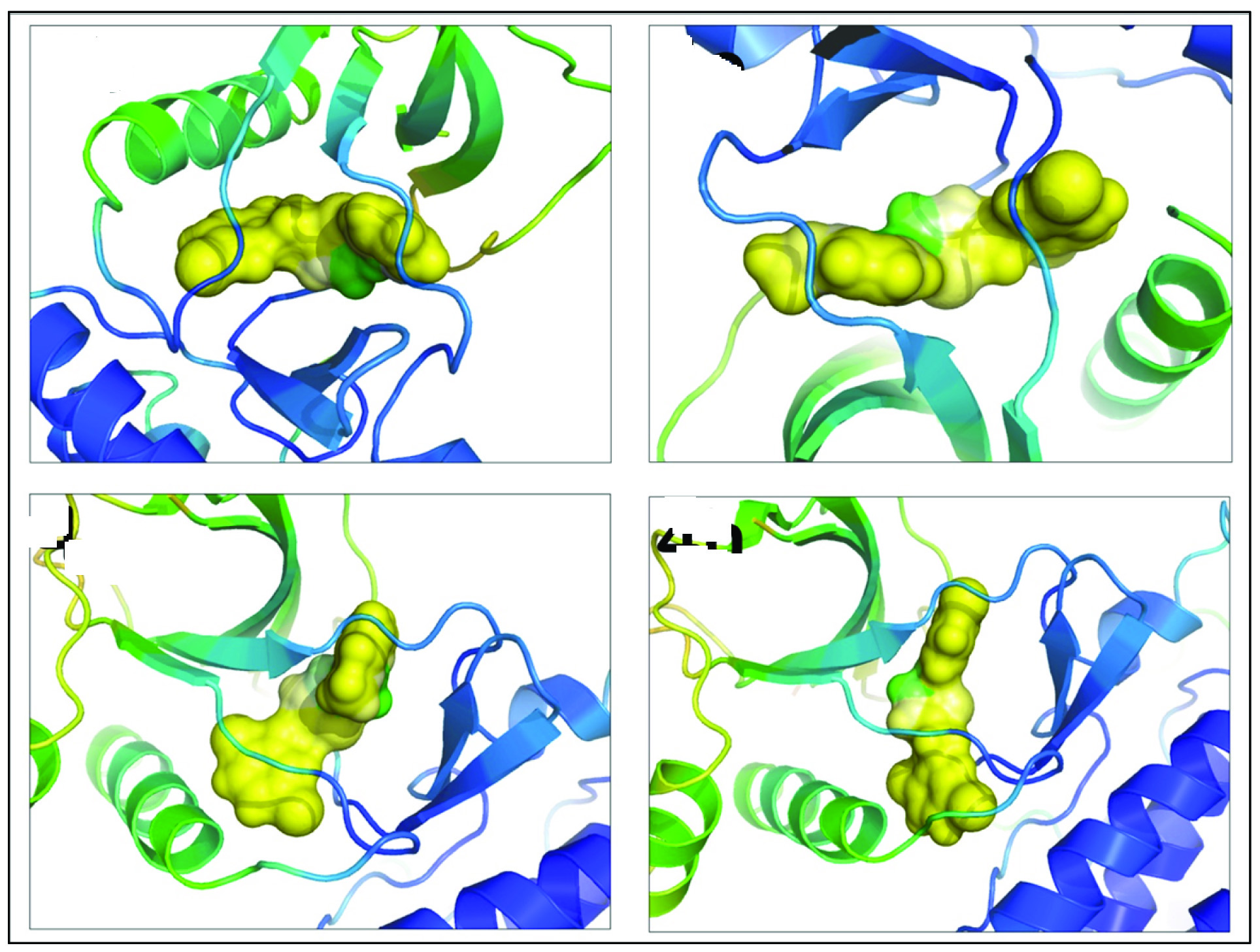
Compound 4b has 92.54% of its surface area deeply buried inside protein, 47.37% is observed to be paired with hydrophobic residues in protein and only 0.27% is revealed to hydrophilic area. These figures clearly indicate greater influence of hydrophobic forces in stabilization of compound 4b-c-Met complex. Thiadiazole derivative 4e, similarly retains one of the highest percentage of buried surface area (93.38%), while its 48.01% of surface area is exposed to hydrophobic residues in ligand binding pocket and only 0.52% area is disclosed to hydrophilic portions. As in otherwise, compound 4g shows maximum percentage of total surface area covered inside c-Met receptor (93.59%) and 54.75% of matching lipophilic surface is observed to be in contact with hydrophobic surface of receptor leaving only 0.67 % of its total surface area in contact with hydrophilic environment. Compound 4h displayed minimal recorded exposure to hydrophilic surrounding (0.15%) and possess highest amount of hydrophobic surface in contact with hydrophobic region of receptor (59.35%) with 90.80% of its total surface area covered inside protein. Comparing relative hydrophobicity coefficients, compound 4h seems to possess highest relative hydrophobicity (0.74) followed by compound 4g (0.71). Thiadiazole compound 4b scored lowest relative hydrophobicity score of 0.64 followed by 4e (0.65). Additionally, calculated MHP data can be visualized by mapping in yellow-white-green scheme where yellow and green colors resembles hydrophobic and hydrophilic characteristics, respectively; thus precisely describing hydrophobic contribution of thiadiazole compounds to c-Met receptor [Table/Fig-10]. [Table/Fig-10] clearly indicate that in case of all promising thiadiazole compounds (i.e., 4b, 4e, 4g and 4h) only the nitrogen atoms of central thiadiazole ring is exposed to hydrophilic environment and rest of the molecule remains deeply buried inside the protein. Thus we can clearly define that central thiadiazole ring might play a decisive role in defining hydrophilic potential of synthesized compounds while terminal di-chloro phenyl and substituted benzyl groups acts imperatively in hydrophobic role of ligand.
Illustration representing mapping MHP data on molecular surface of respective thiadiazole derivatives (surface representation) and c-Met receptor is shown as cartoons.
Conclusion
Here we report effective synthesis of novel 2, 5-disubstituted-1, 3, 4-thiadiazole derivatives. Detailed pharmacophore mapping analysis carried out in this work successfully identifies hepatocyte growth factor receptor as potential target for synthesized derivatives. Moreover, we also identify crucial pharmacophore features that might prove to be a springboard to accelerate research on identification of thiadiazoles scaffolds as novel c-Met inhibitors. Binding free energy values obtained from docking investigation indicate that compounds 4b, 4e, 4g and 4h can be potentially used as lead compounds for c-Met inhibition. By performing detailed structural analysis of docked complexes we revealed the possible interactions with key and conserved residues like Ile-1084, Ala-1108, Val-1092, Lys-1110, Glu-1127, Met-1131, Leu-1140, Val-1155, Leu-1157, Pro-1158, Tyr-1159, Met-1160, Phe-1200, Met-1211, Val-1220, Ala-1221, Asp-1222, Phe-1223 and Met-1131 from c-Met receptor that thiadiazole derivatives are expected to form. On the basis of MHP data we can also expect that hydrophobic forces might play a significant role in thiadiazole dependent c-Met inhibition.