A Case Series of Young Patients with Completely Reversed Severe Pulmonary Hypertension
Aniketh Vijay Balegadde1, Vikrant Vijan2, Rajesh Thachathodiyl3
1 Resident, Department of Cardiology, Amrita Institute of Medical Sciences, Amrita Vishwa Vidyapeetham University, Kochi, Kerala, India.
2 Consultant, Department of Cardiology, Amrita Institute of Medical Sciences, Amrita Vishwa Vidyapeetham University, Kochi, Kerala, India.
3 Professor, Department of Cardiology, Amrita Institute of Medical Sciences, Amrita Vishwa Vidyapeetham University, Kochi, Kerala, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Aniketh Vijay Balegadde, Resident, Department of Cardiology, Amrita Institute of Medical Sciences, Amrita Vishwa Vidyapeetham University, Kochi-682041, Kerala, India.
E-mail: aniketh.vijay7@gmail.com
Pulmonary Arterial Hypertension (PAH) is a progressive symptomatic disorder, which may ultimately lead to death if left untreated. Although majority of PAH cases are idiopathic, pulmonary hypertension resulting due to certain underlying conditions are also observed frequently. In such cases, it becomes essential to identify any potentially treatable or reversible causes for PAH. There have been significant advances in the medical management of PAH and various medicines have been approved by US Food and Drug Administration (FDA) for various stages of PAH. With these therapies, there can be varying degrees of improvement in the pulmonary artery pressures and hemodynamic profile. Therefore, physiologic reversal can and does occur, sometimes to the point of normalization. We hereby present three such cases of severe PAH in patients below 50 years of age due to various aetiologies like left heart disease, isolated unilateral absence of right pulmonary artery with hypoplastic right lung and factor V Leiden mutation associated pulmonary thromboembolism, all of whose pulmonary artery pressures are completely normalised with adequate treatment of the underlying disease and with optimised medications for PAH, ultimately leading to tapering and stoppage of PAH medications.
Factor V Leiden, Hypokinesia, Hypoplastic right lung, Left ventricle, Medical management, Pulmonary artery pressure, Pulmonary thromboembolism, Right ventricle
Case 1
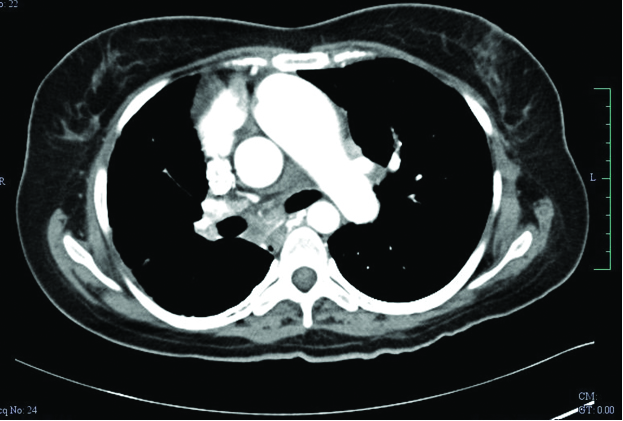
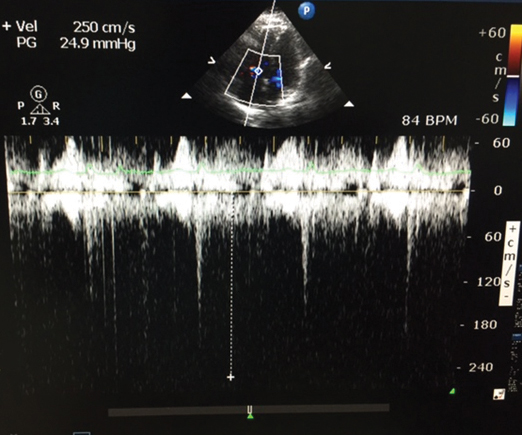
A 28-year-old female, presented to the outpatient department with history of dyspnoea on exertion (Class III, NYHA) since the last three months. She also had a past history of Patent Ductus Arteriosus (PDA) ligation, following which she was totally asymptomatic until three months back. Her body mass index was 24 kg/m2. Clinical examination indicate signs of right heart failure. Chest x-ray revealed decrease in right lung volume and echocardiography revealed severe PAH and non visualisation of right pulmonary artery with main pulmonary artery and left pulmonary artery being dilated. The mean pulmonary artery pressure on admission was 59 mmHg. Right ventricular function was assessed using standard echocardiographic methods, i.e., right ventricular basal tissue Doppler imaging and tricuspid annular plane systolic excursion. Multidetector Computed Tomography (MDCT) of chest was done which confirmed isolated unilateral absence of right pulmonary artery with hypoplastic right lung [Table/Fig-1]. Other causes of PAH were ruled out by necessary investigations. The patient was started on diuretics and sildenafil as per recommended dosage. The dose of sildenafil was gradually tapered every six months based on her pulmonary artery pressures and finally it was stopped once pulmonary artery pressures normalised. In brief, the patient received frusemide 40 mg t.i.d. intravenously on admission, frusemide 40 mg b.i.d. orally on discharge, and the same dose was continued on follow up. On the other hand, the dose of sildenafil was 25 mg t.i.d. initially for one year. Subsequently, it was reduced to 25 b.i.d on regular follow up as pulmonary artery pressures reduced [Table/Fig-2]. The mean pulmonary artery pressure at four year follow up was 27 mmHg. Currently, she is not on any medications for PAH and her pulmonary artery pressure and right ventricular function remain normal.
Case 2
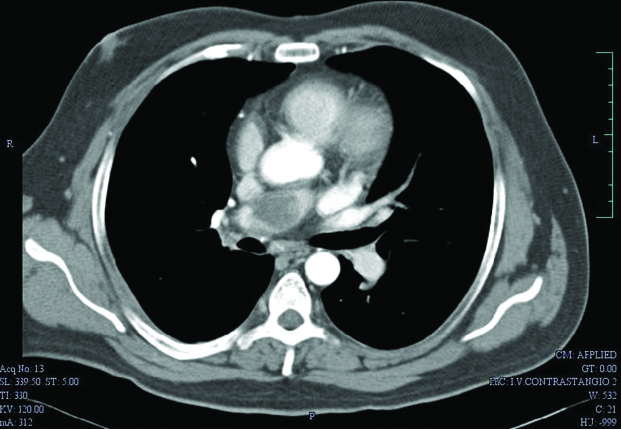
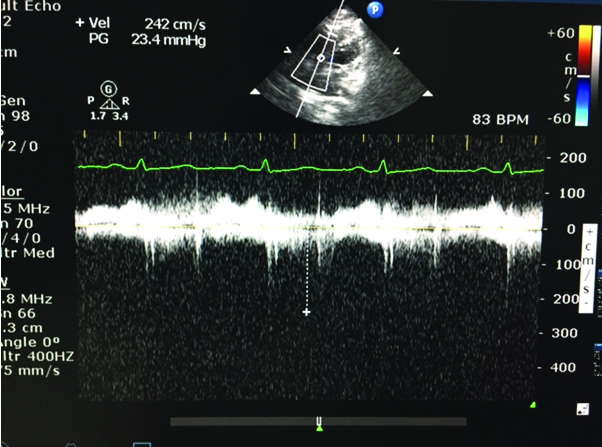
A 48-year-old male, presented to the emergency department with history of acute onset dyspnoea (Class IV, NYHA) for the last two days. On clinical examination, he had a heart rate of 120 bpm, regular in rhythm and normal in volume, blood pressure of 90/50 mmHg and tachypnoeic. Cardiovascular examination revealed palpable P2, fixed narrow splitting of S2, and he had a resting saturation of 80%. An emergency bedside screening echocardiography revealed dilated right atrium/right ventricle, dilated main pulmonary artery, severe PAH and right ventricular dysfunction [Tricuspid Annular Plane Systolic Excursion (TAPSE)-1.0]. A CT pulmonary angiogram was done which confirmed pulmonary thromboembolism involving right main pulmonary artery extending into the lobar arteries [Table/Fig-3]. He was then thrombolysed with streptokinase. On further evaluation, he was found to have factor V Leiden mutation. Other causes of PAH were ruled out by necessary investigations. The patient was treated with oral anticoagulants initially. Sildenafil was started when the pulmonary artery pressure had not reduced even at the end of one month treatment and was continued for three months. At four month follow up, he persisted to have severe PAH with repeat CT pulmonary angiogram showing no evidence of thrombosis. Then, ambriesentan was added to the regime. Both sildenafil and ambriesentan were continued for 20 months. Subsequent follow up at regular interval showed gradual reduction in PA pressures. Thus, sildenafil dose was tapered on regular follow ups and was eventually stopped at 20 follow up, while ambriesentan was stopped as pulmonary artery pressures were normalised at 36 months [Table/Fig-4]. Currently, the patient is not on any medication for PAH, with pulmonary artery pressures and right ventricle function being normal.
Multidetector computed tomography (MDCT) of chest showed isolated unilateral absence of right pulmonary artery with hypoplastic right lung (for Case 1);
Echo image after the treatment (for Case 1).
Computed tomography (CT) pulmonary angiogram showed presence of thrombus in the proximal right pulmonary artery causing near total lumen obstruction (for Case 2);
Echo image after the treatment (for Case 2);
Case 3
A 45-year-old male patient with diabetes mellitus and coronary artery disease who had a past history of inferior wall ST elevation myocardial infarction (five years back) and anterolateral non ST elevation myocardial infarction (two years back). His coronary angiogram revealed diffusely calcified triple vessel disease. He was advised for Coronary Artery Bypass Grafting (CABG), but was unwilling for the procedure. He was kept on medical management. His echocardiography revealed global left ventricular hypokinesia, severe left ventricular systolic dysfunction, grade III diastolic dysfunction, severe PAH, and right ventricular dysfunction. Left heart disease (post-capillary cause) was considered as the major aetiology for PAH. Other causes of PAH were ruled out by necessary investigations. The patient was kept on optimal medical management for coronary artery disease along with tadalafil and ambriesentan for PAH. The other medications used along with PAH medications were dual anti platelets, statins, beta blockers, nitrates, diuretics, aldosterone receptor antagonists, antacids, laxatives. There were no identifiable drug-drug interactions. Subsequent follow up at regular interval showed gradual reduction in pulmonary artery pressures and gradual improvement in right ventricular function, but moderate-to-severe left ventricular systolic dysfunction and diastolic dysfunction still persisted. An advanced echo was done recently, which revealed complete normalisation of pulmonary artery pressures and satisfactory right ventricular function [Table/Fig-5]. He was continued on tadalafil and ambriesentan. Subsequently, the patient underwent orthostatic cardiac allograft transplantation at our institute and is currently recovering well.
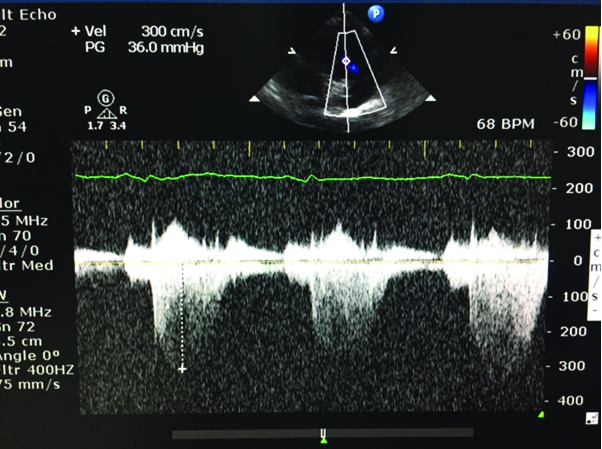
Echo image after the treatment (for Case 3).
Discussion
PAH is a serious and progressive disease of the pulmonary vasculature that can be triggered by a number of known (genetic mutations, toxins, infections or other diseases) and unknown causes [1]. The pathogenesis of PAH is multifactorial, comprising an involvement of various biochemical pathways and cell types. However, it should be noted that all types of PAH, irrespective of underlying aetiology, exhibits pulmonary vasculopathy that can be characterized by intimal proliferation, medial hypertrophy, and development of plexiform lesions [2]. The management of patients with PAH is aimed at two types of remodelling: (a) increased muscularisation of vessel walls as a major cause of pulmonary vascular disease; and (b) Endothelial Cell (EC) proliferation as a cause of pulmonary vascular disease [3]. Here, vascular remodelling that develops because of phenotypically altered EC may be irreversible [2]. Earlier, several studies have reported a strong association between irreversible PAH in congenital heart disease and impaired apoptotic regulation of EC, endothelial damage, and elevated levels of circulating EC [3-5]. On the other hand, the vascular remodelling that develops predominantly because of increased muscularisation of vessel walls is indicated to be reversible. Here, cellular plasticity plays major role in the Smooth Muscle Cells (SMC) shift between a proliferative and a non proliferative phenotype [3]. It remains clinically challenging to convert the phenotypically altered and hyperproliferative SMC to a normal SMC in PAH patients. Various drugs that block abnormal vasoconstriction and inhibit the growth of normal SMC (i.e., endothelin-receptor antagonists, phosphodiesterase type 5 inhibitors, and prostacyclin derivative) are currently used to for the treatment of PAH. It is warranted to develop novel drugs targeting pulmonary vascular remodelling [3].
Previously, various cases have been published wherein the PA pressures have returned to normalization after adequate treatment of the underlying aetiology. Nevertheless, cases where the underlying aetiology is persisting but PA pressures have normalized with adequate treatment (with PAH medications), eventually leading to normalization of PA pressures and stoppage of PAH medications, have not been described to the best of our knowledge. Although treatable causes may be rare in PAH patients, it is important to identify any potential treatable or reversible causes for PAH in these patients [6].
Conclusion
There have been substantial advances in the medical management of PAH and studies demonstrating the significant efficacy and safety of several medicines in treating various stages of PAH. These therapies can be valuable management options to improve PA pressures and hemodynamic profile. It should be acknowledged that physiologic reversal can be attained, even to the point of normalization, in certain PAH cases.
[1]. Humbert M, Sitbon O, Simonneau G, Treatment of pulmonary arterial hypertensionN Engl J Med 2004 351:1425-36. [Google Scholar]
[2]. Howard LS, Prognostic factors in pulmonary arterial hypertension: assessing the course of the diseaseEur Respir Rev 2011 20:236-42. [Google Scholar]
[3]. Sakao S, Tatsumi K, Voelkel NF, Reversible or irreversible remodeling in pulmonary arterial hypertensionAm J Respir Cell Mol Biol 2010 43(6):629-34. [Google Scholar]
[4]. Lévy M, Maurey C, Celermajer DS, Vouhé PR, Danel C, Bonnet D, Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart diseaseJ Am Coll Cardiol 2007 49:803-10. [Google Scholar]
[5]. Smadja DM, Gaussem P, Mauge L, Israël-Biet D, Dignat-George F, Peyrard S, Circulating endothelial cells: a new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart diseaseCirculation 2009 119:374-81. [Google Scholar]
[6]. Kapoor R, Khan A, Barrantes J, Ross R, Gowda S, Saydain G, A rare potentially reversible cause of severe pulmonary hypertension in a young adult hypoxic patientChest 2011 140:177A [Google Scholar]