Unusual Presentation of Atypical Infantile Pompe Disease in the Newborn Period with Left Ventricular Hypertrophy
Sanjay Kumar1, Amit Kumar2
1 Senior Resident, Department of Paediatrics, AIIMS, New Delhi, India.
2 Senior Resident, Department of Paediatrics, VMMC and Safdarjung Hospital, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Amit Kumar, Jyoti Singh, 140 G, IInd Floor, Gautam Nagar, New Delhi-110049, India.
E-mail: amitofpmch@gmail.com
Pompe disease, also known as glycogen storage disease Type II, is a lysosomal storage disorder caused by α-glucosidase deficiency. In general, the clinical spectrum varies with respect to the age of onset, residual enzyme activity and organ involvement. Infantile onset disease has two subtypes: classical and non-classical (atypical). This case report describes the case of a newborn who presented with generalized hypotonia and elevated serum enzyme levels of aspartate aminotransferase 93 IU/L, lactate dehydrogenase 888 IU/L and creatine kinase 670 μg/L. The electrocardiogram showed short PR interval with large QRS complexes with echocardiography suggesting evidence of left ventricular hypertrophy with infiltration in its walls. On the basis of the clinical signs and laboratory results, dried blood spots from the baby were tested to determine the acid α-glucosidase (GAA) activity, and the result confirmed that the GAA activity was only 1.42 units, normal range 5.5 to 29.6 units, leading to a diagnosis of Pompe disease (atypical infantile). Recognizing this disease and initiating enzyme replacement therapy in infants at the earliest can improve the quality of life of patients.
Enzyme replacement therapy, Lysosomes, Non-classical (atypical)
Case Report
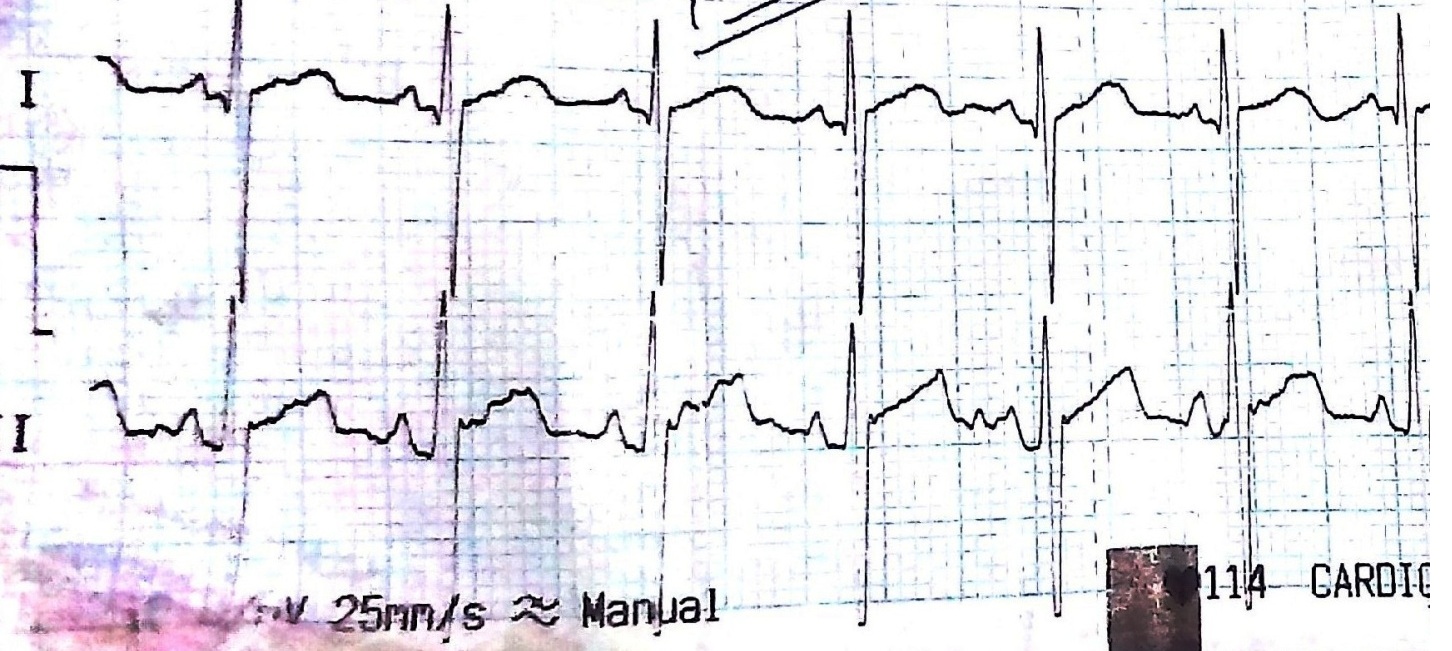
A full term female newborn was born after spontaneous vaginal delivery with uneventful antenatal and natal period. She was born of non-consanguineous marriage, 3rd in order, the other two siblings were alive and healthy. She weighed 4 kg with length of 51 cm and had no gross congenital anomalies detected at birth, so was rooming-in with the mother. At 20 hours of life, baby developed tachypnea (RR) 70/minutes with mild subcostal retractions (Downe’s score: 2). Baby was admitted in neonatal intensive care unit and vital parameters measured which were within normal limits. Detailed systemic examination revealed generalized hypotonia without any dysmorphic features, macroglossia or hepatomegaly. At 36 hours of life, respiratory distress increased further requiring increased oxygen supplementation and thereafter, ventilatory support. Sepsis work up and chest X-ray were found to be normal [Table/Fig-1]. Possibility of spinal muscular dystrophy/congenital muscular dystrophy/congenital myopathy was kept. Biochemical analysis showed increased levels: alanine aminotransferase (ALT) 70 IU/L, aspartate aminotransferase (AST) 93 IU/L, Lactate Dehydrogenase (LDH) 888 IU/L and creatinine phosphokinase (CPK) 670 μg/L. The ratio of CPK to transaminase level was greater than 2, so pompe disease was considered in the differential as well. The electrocardiogram showed sinus rhythm 140 per minute, short PR interval (0.04 sec) and tall and large QRS complexes pointing more towards pompe disease due to coexistence of myopathy with cardiomyopathy [Table/Fig-2]. Baby got subsequently weaned off from ventilator. Transthoracic echocardiography revealed significant left ventricular hypertrophy with infiltration in its walls with absence of obstruction across the outflow tract. The posterior and septal wall thickness of left ventricle was 6 mm and 7 mm respectively, >2 SD above normal for age with other findings being normal. The Dried Blood Spot analysis (DBS) for α-glucosidase activity level in leucocyte was reduced (1.42 units, normal range 5.5 to 29.6 units) confirming the diagnosis. So, ERT (20 mg/kg) every alternate week was started from six weeks of age. On follow up examination at 12 months of age, she weighed 10 kg with length of 78 cm, was able to stand with support and walk holding furnitures without any episode of acute respiratory insufficiency. Present echocardiographic findings revealed normal septal and posterior wall thickness of left ventricles 6 mm and 5 mm respectively. The dimensions of left ventricle, diastolic and systolic were 24 mm and 17 mm with fractional shortening of 30%. Up till now, there is no significant side effect of ERT.
Chest X-ray showing normal lung fields and cardiac silhouette.
ECG showing characteristic short PR interval and large QRS complex as present in Pompe disease.
ERT for pompe disease is known as “MYOZYME (alglucosidase alfa), a lysosomal glycogen-specific enzyme, consists of the human enzyme acid α-Glucosidase (GAA), encoded by the most predominant of nine observed haplotypes of this gene. It is produced by recombinant DNA technology in a Chinese hamster ovary cell line. The cost per year varies according to the weight and ranges from 35 lacs to 75 lacs per year till 20 kg. It can be procured after prior order to pharmaceutical company.
Discussion
Pompe disease is a rare inherited autosomal recessive lysosomal storage disorder. It occurs due to deficiency of acid α-glucosidase (acid maltase) enzyme in the lysosomes resulting in impaired degradation of glycogen into glucose.
Pompe disease is broadly classified into infantile (classical and atypical) and late onset (juvenile and adult onset) forms. The classical infantile form usually presents soon after birth with marked muscle weakness and hypertrophic cardiomyopathy. They often succumb to death in the first year of life due to cardio respiratory failure [1]. Atypical infantile Pompe disease generally presents few months later with slower progression and less severe cardiomyopathy. The mean age of presentation in atypical infants is 4.8 ± 2.9 months [2]. These patients usually develop left ventricular hypertrophy after age of six months without any outflow obstruction [3].
With the advent of Enzyme Replacement Therapy (ERT), classical infantile and late-onset pompe disease have improved survival, cardiorespiratory function, growth and developmental milestones. However, paucity of data exists in the literature mentioning the modified natural course in atypical infantile pompe disease after ERT.
Pompe’s disease, also known as glycogen storage disorder Type II is a genetic muscular disorder caused due to deficiency of α-glucosidase enzyme. The overall reported incidence varies from one in 14,000 to one in 300,000 depending on the geographic area or ethnic group [4]. The gene encoding enzyme (GAA) is located on chromosome 17q25.2-q25.3. More than 200 mutations have been reported so far [5]. There is wide variety of clinical presentation ranging from the classical form with early onset and severe phenotype to non-classical form with later onset and milder phenotypes.
The classical infantile form presents with feeding problem, difficulty in sucking and swallowing resulting in poor weight gain and frequent respiratory tract infections. Myopathy is quite universal with diaphragm, lower limb and trunk muscles more severely affected. About 50% of infants have either macroglossia, moderate hepatomegaly or both. Glycogen gets accumulated in smooth and striated muscles, in the liver and in the motor neurons of spinal cord and brainstem leading to progressive deterioration and death at seven months of age [6]. Most infants develop massive and progressive cardiomegaly before six months of age. Glycogen accumulation in cardiac muscle causes thickening of the walls of both ventricles and interventricular septum, resulting in a hypertrophic cardiomyopathy, which progresses to a dilated cardiomyopathy. Increasing left ventricular thickness can also lead to obstruction of the left ventricular outflow tract.
In contrast, late-onset Pompe disease can present at any time from the toddler years to adulthood without any significant cardiac manifestations, slower progression of muscle disease, and a less severe prognosis than infantile-onset cases.
There is a subtype of classical infantile pompe disease referred as ‘atypical’ by Slonim AE et al., [2]. Atypical infantile Pompe’s diseases usually have similar presentation but the onset of disease occurs at a later age developing left ventricular hypertrophy without outflow obstruction after six months of age. The electrocardiographic findings and serum creatine kinase, aspartate aminotransferase, and alanine aminotransferase levels in the classical and non-classical infantile forms are usually similar. Acid α-glucosidase enzyme activity was undetectable in typical infants, whereas traces of activity (usually <0.5%) were detected in non-classical infants. Such infants usually develop acute respiratory insufficiency requiring assisted ventilation and need supportive feeding to improve weight gain.
Conclusion
The above case report was unique in its presentation with respiratory distress being the prominent symptom with left ventricular hypertrophy present in the neonatal period. A high index of suspicion for pompe disease should always be considered in neonates presenting with generalized hypotonia to prevent missing atypical cases. Appropriate early treatment help to achieve normal growth, motor development, cardiac indices and prevent from frequent respiratory infections.
[1]. Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case L, Pompe disease diagnosis and management guidelineACMG Practice Guidelines 2006 8(5):267-88. [Google Scholar]
[2]. Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F, Identification of two subtypes of infantile acid maltase deficiencyJ Pediatr 2000 137:283-85. [Google Scholar]
[3]. Kishnani PS, Howell RR, Pompe disease in infants and childrenJ Pediatr 2004 144:S35-S43. [Google Scholar]
[4]. Hirschhorn R, Reuser AJJ, Glycogen storage disease type II: Acid alphaglucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, et al, edsThe metabolic and molecular bases of inherited disease 2001 New YorkMcGraw Hill:3389-420. [Google Scholar]
[5]. Manganelli F, Ruggiero L, Clinical features of Pompe diseaseActa Myologica 2013 32(2):82-84. [Google Scholar]
[6]. Samuel Ignacio Pascual, Phenotype variations in early onset pompe disease: Diagnosis and treatment results with myozyme C. Espinós et al. (eds.)Inherited Neuromuscular Diseases, Advances in Experimental Medicine and Biology:652 [Google Scholar]