Detection of Compound Heterozygous Sickle Cell-β+ Thalassaemia in a Patient with Extreme Weakness, Mild Jaundice and Moderate Anaemia - A Case Report
Subhash Chandra1, Mostafa Ali2, Pooja Mishra3, Ashok Kumar Kapoor4, Yamini Jindal5
1 Professor, Department of Pathology, GCRG Medical College, Lucknow, Uttar Pradesh, India.
2 Professor, Department of Pathology, Vivekanand Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
3 Senior Technician, Department of Pathology, Vivekanand Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
4 Pathologist, Department of Pathology, RML Mehrotra Pathology Pvt Ltd, Lucknow, Uttar Pradesh, India.
5 Resident, Department of Pathology, RML Mehrotra Pathology Pvt Ltd, Lucknow, Uttar Pradesh, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Ashok Kumar Kapoor, RML Mehrotra Pvt. Lmt, Nirala Nagar, Lucknow-226020, Uttar Pradesh, India.
E-mail: drashokkapoor2016@gmail.com
A 16-year-old female complained of extreme weakness. She had moderate anaemia; her Haemoglobin (Hb) was 7.7 gm/dl. Peripheral blood smear showed few sickled red cells. Sickle cell test was positive. High-Performance Liquid Chromatography (HPLC) revealed elevated levels of HbS (38.4%) and HbF (15.7%). In addition, HbA2 concentration was 3.8% and HbA concentration was 42.1%. Results suggested a diagnosis of compound heterozygous sickle cell-β+ thalassaemia. Sickle cell test was also positive with blood of patient’s brother; Hb HPLC examination showed relatively low concentration of HbS (25.2%) suggesting a diagnosis of sickle cell trait. Chromatogram of patient’s step sister suggested a diagnosis of thalassaemia trait. Findings of this study suggested that abnormal genes were inherited in the patient from both the parents. It was interpreted that repeated haemolysis in the patient might have contributed to anaemia, weakness, rise in indirect bilirubin and jaundice. Furthermore, high level of HbF (>12%) may interfere with polymerization of sickle haemoglobin suggesting beneficial effects of HbF-inducing agents which may inhibit sickling.
Bilirubin, Defective globin chain synthesis, Gene, High-performance liquid chromatography
Case Report
A female patient, 16-year-old complained of severe generalized weakness for last two years. She belonged to Tharu-Chaudhary community of Nepal. She had mild jaundice. Spleen was not palpable on abdominal examination. Complete blood examination revealed moderate microcytic hypochromic anaemia (Hb 7.7 gm/dl). Few sickle cells (3.0%), anisocytosis and plenty of late normoblasts (6%) were seen. Sickle cell test was positive; it is a slide-based test for sickling with sodium metasulphite. Severe neutrophilia was seen (absolute neutrophil count was 31300 cells/mm3). Platelet count was 0.68 lac/mm3 suggesting thrombocytopenia. Serum iron was 236 μg/dl (normal 50-150 μg/dl). Serum total iron binding capacity was 347 μg/dl (normal 250-450 μg/dl). Serum ferritin was >1500 ng/ml (normal 13-150 ng/ml); it suggested iron overload. Serum albumin was 3.9 gm/dl. Total serum bilirubin was 3.6 mg/dl. Indirect serum bilirubin was 2.66 mg/dl, suggesting haemolysis. She was given daily supplement of folic acid and desferrioxamine s.c.i.
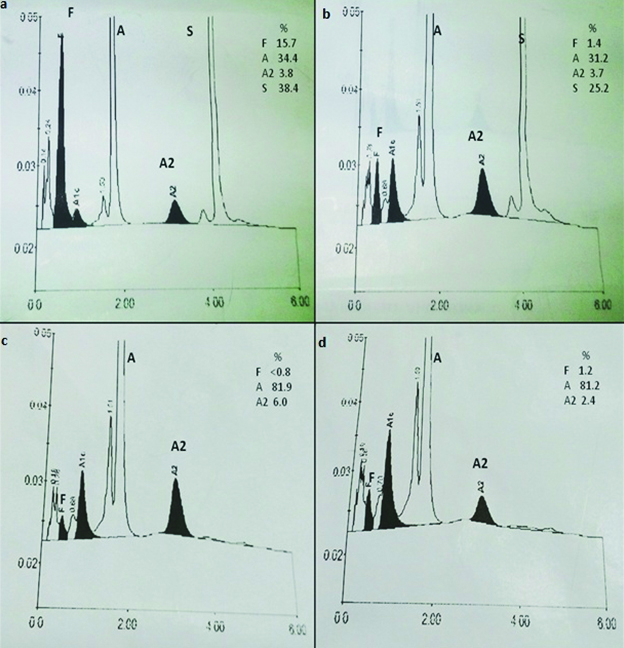
Ion-exchange HPLC was done. Extended program mode was adjusted using Bio-Rad D10 (Bio-Rad laboratories Inc. Hercules, CA 94547). It was used for separation of different haemoglobins including Hb variants based on their ionic interactions with the cartridge material. Positive and negative controls were also run with patient’s sample. Result of Hb HPLC of patient revealed presence of high levels of HbS (38.4%) and HbF (15.7%); HbA2 concentration was 3.8%. In addition, HbA concentration was 42.1%. Hb HPLC findings suggested a diagnosis of double heterozygous sickle cell-β+ thalassaemia [Table/Fig-1a]. Blood samples were also collected from patient’s brother and step sister. Sickling test was positive with blood of patient’s brother; his total Hb was 15.4 gm/dl. Hb chromatogram suggested a diagnosis of sickle cell trait [Table/Fig-1b]. [Table/Fig-1c] shows the result of Hb HPLC with patient’s sister. Her chromatogram suggested a diagnosis of β+ thalassaemia trait. [Table/Fig-1d] shows chromatogram of a normal unrelated subject.
(a) shows results of haemoglobin HPLC in a patient with HbS-β+ thalassaemia. Extended mode shows HbS peak at retention time of 4.2 min.
(b) Chromatogram of brother of above patient. Result of HbHPLC suggested a diagnosis of sickle cell trait. It shows a prominent peak in the range of HbA2 (6.0%).
(c) Result of Hb HPLC with blood of patient’s step sister. Findings suggested a diagnosis of β+ thalassaemia trait. It showed a prominent peak in the range of HbA2 (6.0%).
(d) Normal Hb HPLC from an unrelated subject.
X axis = type of Hb; Y axis = Amount of Hb.
She was given daily supplements of folic acid and deferrioxamine s.c.i. Later she could not be followed further.
Discussion
HbS gene was first detected in India in an Irula boy in Nilgiri hills [1]. Later, this pathologic gene was found in sickle cell belts of central India [2]. A patient with sickle cell-β thalassaemia has been reported earlier from Nepal [3]. Rarely, cases of double heterozygous HbS-β+ thalassaemia have been reported from Kuwait [4] and India [5]. In another study, 12 cases of double heterozygous for HbS and beta thalassaemia minor (SB+) were detected [6].
Interesting finding of this case was the detection of moderate anaemia in a patient with HbS-β+ thalassaemia. Anaemia appeared to have developed following frequent mild haemolysis followed by mild rise in indirect bilirubin and jaundice. In addition, the patient had high level of HbF (15.7%). HbF formation has been shown to improve the morbidity pattern of the patients with sickle cell disease [7].
Two types of HbS-β thalassaemia have been described, i.e., with HbA (HbS-β+ thalassaemia) and without HbA (HbS-βo thalassaemia). Both types have distinctive hematological and clinical features. HbS-βo thalassaemias have lower Hb levels, a more rapid haemolysis and more severe course than HbS-β+ thalassaemia types [8]. In HbS-β+ thalassaemia, variable amounts of HbA dilute HbS and inhibit Hb polymerization-induced cellular damage. The HbA levels may vary from <5% to 45% of the haemolysate and higher levels of HbA are usually associated with a milder phenotype [9]. In the current case, HbA concentration was 42.1%; this concentration might have reduced the morbidity of current case. However, occasional patient with compound heterozygous state may not develop symptoms until the onset of puberty, early adult life or pregnancy [10]. Genes modifying the inflammatory response or expression of cytokines may alter the clinical severity of disease [10]. Furthermore, molecular studies revealed that among three mutations causing HbS-β+ thalassaemia type III, levels of HbA2, HbF, MCV and MCH were highest in the – 88 and lowest in poly A mutations [11]. Clinically, HbS-βo thalassaemia and HbS-β+ thalassaemia type I were generally severe and HbS-β+ thalassaemia type III disease with – 88 mutation was milder than that caused by poly A mutation [11].
Conclusion
Proper diagnosis of patients with sickle cell disease is important. In absence of a correct diagnosis, the patients are given iron supplements which further increase the iron overload. Both HbF and HbA production appeared to improve the prognosis of a patient with sickle cell-β+ thalassaemia when compared with sickle cell-βo thalassaemia. Further, it suggested possible role of HbF-inducing agents in the treatment of HbS-β+ thalassaemia.
Ethical consideration: Written consent was taken from all the subjects included in this study.
[1]. Lehman H, Cutbush M, Sickle cell trait in South IndiaBritish Med Journal 1952 1(4755):404-05. [Google Scholar]
[2]. Urade BP, Incidence of sickle cell anaemia and thalassaemia in central IndiaOpen J of Blood diseases 2012 2:71-80. [Google Scholar]
[3]. Adhikari RC, Shreshtha TB, Shreshtha RB, Subedi RC, Parajuli KP, Dali S, Sickle cell disease-case reportsJ of Nepal Med Association 2003 42:36-38. [Google Scholar]
[4]. El-Hazmi MAF, M-Hazmi A, Warsy AS, Sickle cell disease in middle East Arab countriesInd J Med Res 2011 134(5):597-610. [Google Scholar]
[5]. Mukherjee MB, Nadkarni AH, Gorakshakar AC, Ghosh K, Mohanty D, Colah RB, Clinical, hematological and molecular variability of sickle cell-β-thalassaemia in western IndiaInd J Hum Genet 2010 16(3):154-58. [Google Scholar]
[6]. Panigrahi S, Patra PK, Khodiar PK, The screening and morbidity pattern of sickle cell anemia in ChattisgarhInd J Hematol Blood transfusion 2015 31(1):104-09. [Google Scholar]
[7]. Maude GH, Hayer RJ, Serjeant GR, The hematology of steady state homozygous sickle cell disease: interpretationship between hematological indicesBr J Hematol 1987 66:649-58. [Google Scholar]
[8]. Serjeant GR, Ashcroft MT, Serjeant BE, Milner PF, The clinical features of sickle cell/β-thalassaemia in JamaicaBr J Hematol 1973 24:19-30. [Google Scholar]
[9]. Figueiredo MS, The compound state: HbS/betathalassaemiaRev Bros Hematol Haemoter 2015 37(3):150-52. [Google Scholar]
[10]. Benj EJ, Disorders of haemoglobinIn ‘Harrison’s principles of Internal medicine 2012 18th edMcGraw Hill Medical:852-861.Longo et al [Google Scholar]
[11]. Serjeant GR, Serjeant BE, Fraser RA, Hambleton IR, Higgs DR, Kulozik AE, HbS-β-thalassaemia: molecular, hematological and clinical comparisonsHaemoglobin 2011 35(1):1-12. [Google Scholar]