Waldmann’s Disease (Primary Intestinal Lymphangiectasia) with Atrial Septal Defect
Shrikiran Aroor1, Suneel Mundkur2, Shravan Kanaparthi3, Sandeep Kumar4
1 Professor and Head, Department of Paediatrics, Kasturba Medical College, Manipal, Karnataka, India.
2 Additional Professor, Department of Paediatrics, Kasturba Medical College, Manipal, Karnataka, India.
3 Senior Resident, Department of Paediatrics, Kasturba Medical College, Manipal, Karnataka, India.
4 Assistant Professor, Department of Paediatrics, Kasturba Medical College, Manipal, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Shravan Kanaparthi, Flat 206, Mandavi Sapphire, Perampally Road, Manipal-576101, Karnataka, India.
E-mail: shravan.1354@gmail.com
Waldmann’s disease or Primary Intestinal Lymphangiectasia (PIL) is a rare disorder of gastrointestinal tract characterized by dilated lymphatics and widened villi causing leakage of lymph into intestinal lumen. Loss of lymph leads to hypoalbuminemia, hyogammaglobulinemia and lymphopenia. Secondary lymphangiectasia occurs secondary to an elevated lymphatic pressure as in lymphoma, systemic lupus erythematosus, constrictive pericarditis, cardiac surgeries (Fontan’s procedure), inflammatory bowel disease and malignancies. We, hereby present a five-year-old male child who presented with abdominal distension and poor weight gain. He had hypoalbuminemia, lymphocytopenia and hypogammaglobulinemia. Upper gastrointestinal endoscopy showed normal gastric mucosa and punctate white lesions in duodenal mucosa with biopsy confirming intestinal lymphangiectasia. Secondary causes of intestinal lymphangiectasia were ruled out. Echocardiography revealed atrial septal defect which is an uncommon association with Waldmann’s disease. He was started on low fat, high protein diet and medium chain triglyceride supplementation following which he improved symptomatically. High index of suspicion, early diagnosis and appropriate dietary treatment are necessary to alleviate symptoms as well as to achieve a sustainable growth and development in these children.
Anasarca, Protein loosing enteropathy, Recurrent diarrhoea
Case Report
A five-year-old male child presented with a history of abdominal distension and recurrent episodes of diarrhoea associated with poor weight gain for last two years. There was no history of abdominal pain, vomiting, fever, yellowish discolouration of eyes, difficulty in breathing, rash or urinary symptoms. His weight (14.7 kg with oedema) for age was <3rd centile and height (104.5cm) for age was between 3rd-15thcentile.
Clinical examination revealed anasarca, bilateral pitting pedal oedema, liver with soft consistency (span-8 cm) without splenomegaly. Vitals and other system examination were within normal limits. Investigations revealed hypoalbuminemia (2.1 gm/dL), hypoglobulinemia (1.5 gm/dL), normal liver enzymes and INR (International Normalized Ratio) (1.16), normal counts (5400/cumm) with lymphopenia (13%). Urine evaluation, serum lipid profile, thyroid profile and ceruloplasmin levels were normal.
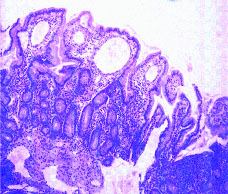
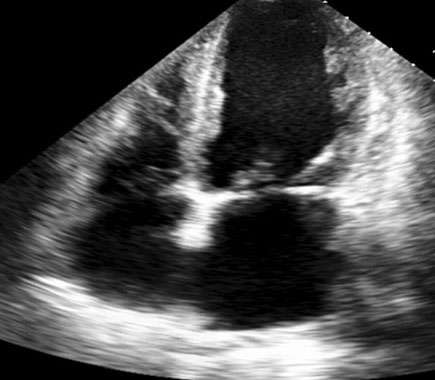
Ultrasonography of abdomen revealed normal liver echotexture and ascites. Ascitic fluid analysis was suggestive of a transudate (protein 1.3 gm/dL). Workup for tuberculosis was negative (acid fast bacillus staining and PCR). Liver biopsy was normal. Upper gastrointestinal endoscopy showed normal gastric mucosa with whitish punctate mucosal spots distributed throughout duodenal mucosa (D2 & D3) suggestive of dilated lacteal vessels. Biopsy of D3 segment was suggestive of intestinal lymphangiectasia [Table/Fig-1]. Immunoglobulin analysis showed low serum IgG levels (<222 mg/dL). Further investigations, to rule out secondary causes of intestinal lymphangiectasia {colonoscopy, colon biopsy, ANA (Antinuclear Antibody) profile, Contrast-Enhanced Computed Tomography (CECT) abdomen, mesenteric lymph node and peritoneal biopsy} were normal. Echo cardiograph showed 9 mm ostium secundum atrial septal defect [Table/Fig-2] with no evidence of constrictive pericarditis.
Duodenal biopsy (H&E stain, 10X) showing bulbous villi with focal blunting lined by columnar epithelium with goblet cells overlying dilated lymphatic channels at tips suggestive of intestinal lymphangiectasia.
Echo-cardiograph showing 9 mm ostium secundum atrial septal defect.
A diagnosis of Waldmann’s disease with atrial septal defect was considered and he was started on protein rich diet, MCT (Medium Chain Triglycerides) supplementation along with LCFA (Long Chain Fatty Acids) restricted diet. He responded to the treatment and improved symptomatically (diarrhoea, anasarca and net weight gain-1.4 kg) over next four weeks.
Discussion
Intestinal lymphangiectasia, a rare disease, is characterised by abnormal intestinal lymphatic system leading to loss of plasma and thereby protein and lymphocyte loss into the gut. Primary and secondary, are the two types of intestinal lymphangiectasia described in literature [1]. In primary lymphangiectasia there is no predisposing condition for increased lymphatic pressures and is probably due to congenital anatomic malformation of the lymphatics. Primary intestinal lymphangiectasia was first described by Waldmann T et al., in 1961, with very rare familial forms reported in literature [2]. Yellow nail syndrome, Klippel-trenaunay-Weber syndrome, Von Recklinghausen, Noonan, Turner and Hennekam are associated with PIL [3-6]. Hyposplenism, thymic hypoplasia and neutrophil dysfunction are few reported associations [4]. Secondary intestinal lymphangiectasia is an acquired disease, characterised by increased intestinal lymph leakage, secondary to increased pressure due to obstruction within the lymphatics. Mesenteric lymphadenopathy, fibrosis of lymph drainage tracts, constrictive pericarditis, inflammatory bowel diseases, systemic lupus erythematosus, tuberculosis, parasitic and bacterial infections, malignancies like lymphomas are the causes of secondary intestinal lymphangiectasia [4,7,8].
PIL is characterised by protein losing enteropathy due to increase in intestinal lymphatic fluid loss. Hypoproteinemia is most often manifested as hypoalbuminemia due to the long turnover rate of albumin [3,4,6-8]. Loss of lymphocytes in the intestine manifests as lypmphocytopenia, which if severe can lead to impaired humoral and cellular immunity. Preserved memory and effector CD4T cells were probably responsible in preventing recurrent infections in PIL [3]. Oedema (symmetric or asymmetric), abdominal distension, poor weight gain, vomiting, recurrent diarrhoea, malabsorption manifesting as fat soluble vitamin deficiencies are the predominant clinical features of PIL [5,6,9].
Diagnosis is suggested by clinical features along with hypoproteinemia, lymphocytopenia. Other causes of hypoproteinemia should be ruled out [3,4,7,8]. Stool analysis may show steatorrhea and increased α anti trypsin clearance (>24 ml/day) [8]. Barium studies may show fluid hypersecretion and nodular or punctate lucencies in mucosa of small bowel. Radionuclide studies using 99mTechnetium-labelled dextran scintigraphy and endoscopy (enteroscopy, capsule endoscopy) helps in localising the lesion and biopsy helps in providing a definite diagnosis. Secondary causes of intestinal lymphangiectasia have to be ruled out before considering it as a PIL. Antinuclear Antibody (ANA) profile, abdominal imaging to rule out significant lymphadenopathy and retroperitoneal fibrosis, peritoneal and lymph node biopsy to look for malignancy and tuberculosis, colonoscopy to look for inflammatory bowel disease and workup for infectious aetiologies should be done in all cases [3,4,6,7].
Diet low in LCFA, high in protein and Medium Chain Triglycerides (MCT) is usually advised [6,10,11]. MCTs are not re-esterified within the intestinal cells and thus bypass the enteric lymphatics and directly enter the portal system. Decreased LCFA decreases the lymphatic flow and pressure in lymphatic system. Supplementation of fat soluble vitamins and calcium salts may be required. Few cases with localised lesions may improve following surgical resection. Octreotide, corticosteroids and antiplasmin can be considered in cases who don’t respond to dietary management [3-5,7,8].
Conclusion
An early diagnosis of intestinal lymphangiectasia assumes great importance in children with recurrent diarrhoea and ansarca. Detailed workup should be done to rule out secondary causes of lymphangiectasia and to detect the other associated abnormalities. Lifelong dietary therapy with high protein and MCT supplementation remains the mainstay of treatment to achieve a sustainable growth and development.
[1]. Wen J, Tang Q, Wu J, Wang Y, Cai W, Primary intestinal lymphangiectasia: four case reports and a review of the literatureDig Dis Sci 2010 55(12):3466-72. [Google Scholar]
[2]. Waldmann T, Steinfeld J, Dutcher T, Davidson J, Gordon R, The role of the gastrointestinal system in idiopathic hypoproteinemiaGastroenterology 1961 41:197-207. [Google Scholar]
[3]. Vignes S, Bellanger J, Primary intestinal lymphangiectasia (Waldmann’s disease)Orphanet Journal of Rare Diseases 2008 3:5 [Google Scholar]
[4]. Proujansky R, Protein losing enteropathy. In: Walker, Goulet, Kleinman, Sherman, Shneider, Sanderson, editorsPaediatric gastrointestinal disease: Pathophysiology, diagnosis and management 2004 4th edOntarioBC Decker:194-199. [Google Scholar]
[5]. Suresh N, Ganesh R, Janani S, Malathi S, Primary intestinal lymphangiectasiaIndian Pediatrics 2009 46(10):903-06. [Google Scholar]
[6]. Marjet JB, Koert MD, Merit MT, Protein-losing enteropathy in childrenEur J Pediatr 2010 169:1179-85. [Google Scholar]
[7]. Soon JL, Hyun JS, Boo S-J, Na S-Y, Heung UK, Chang LH, Primary intestinal lymphangiectasia with generalized wartsWorld Journal of Gastroenterology 2015 21(27):8467-72. [Google Scholar]
[8]. David Greenwald, Protein losing gastroenteropathy. In: Mark F, Friedmann LS, Brandt LJSleisenger & Fordtran’s gastrointestinal and liver disease: Pathophysiology, diagnosis and management 2010 10th edPhiladelphiaElsevier Saunders:464-470. [Google Scholar]
[9]. Oregon V, Mayans R, Bustamante C, Monjaraz T, Mendez C, Leon C, Primary intestinal lymphangiectasia: twenty years of experience at a mexican tertiary care hospitalJournal of Gastroenterology Mexico 2014 79(1):7-12. [Google Scholar]
[10]. Munck A, Sossa V, Faure C, Besnard M, Ferkdadji L, Cezard J, Long-term follow up of primary intestinal lymphangiectasia in the child. Six case reportsArch Pediatr 2002 9(4):388-91. [Google Scholar]
[11]. Greco F, Piccolo G, Sorge A, Pavone P, Triqlia T, Spina M, Early onset of primary intestinal lymphangiectasia. A case report and diet treatmentMinerva Pediatr 2003 55(60):615-19. [Google Scholar]