Introduction
CLOVES syndrome is a rare overgrowth disorder characterized by congenital lipomatous overgrowth, vascular malformation, epidermal nevi and spinal abnormalities. Clinical aspects described by Alomari AL and Sapp JC [1,2] where mostly in patients over one-year-old, whereby, the aim of our report is to describe three fatal cases of newborns with CLOVES syndrome and their clinical features.
The case report is on three patients who were pre-natally diagnosed with masses in the body. All of them were born of unremarkable gestations in which there were no maternal illnesses or teratogenic agents reported. The maternal ages ranged between 23 and 35 years, and all patients were Caucasian.
Case Series
Case-1
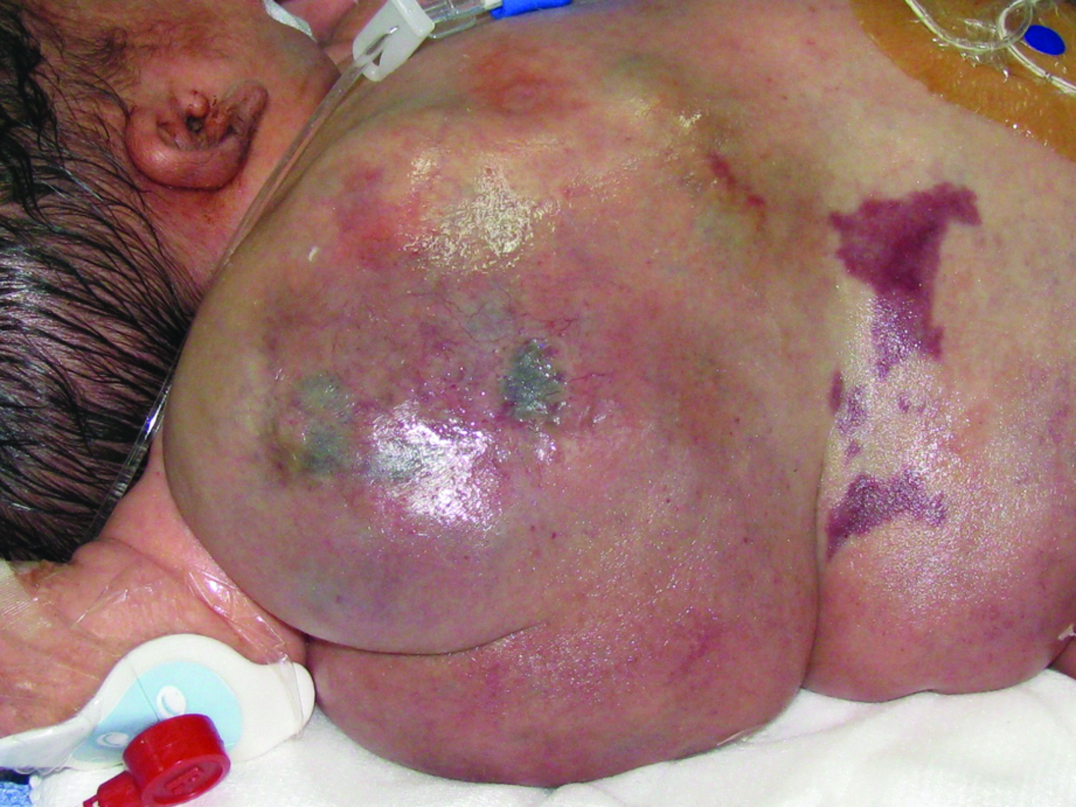
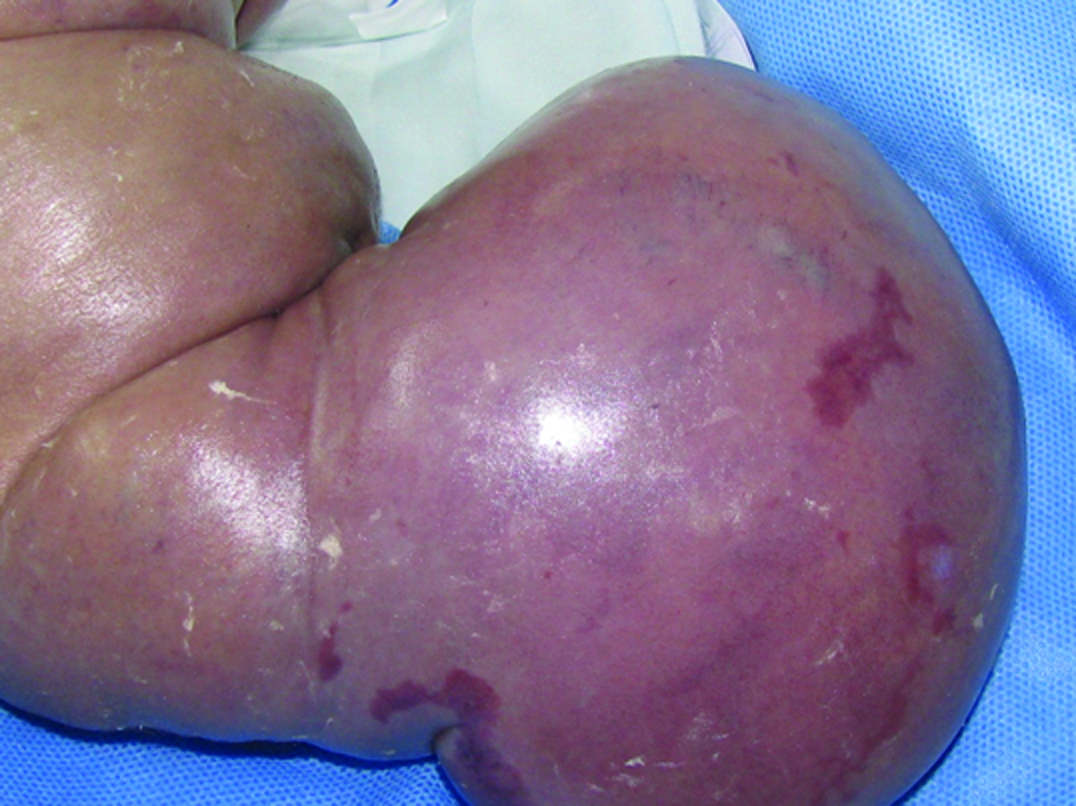
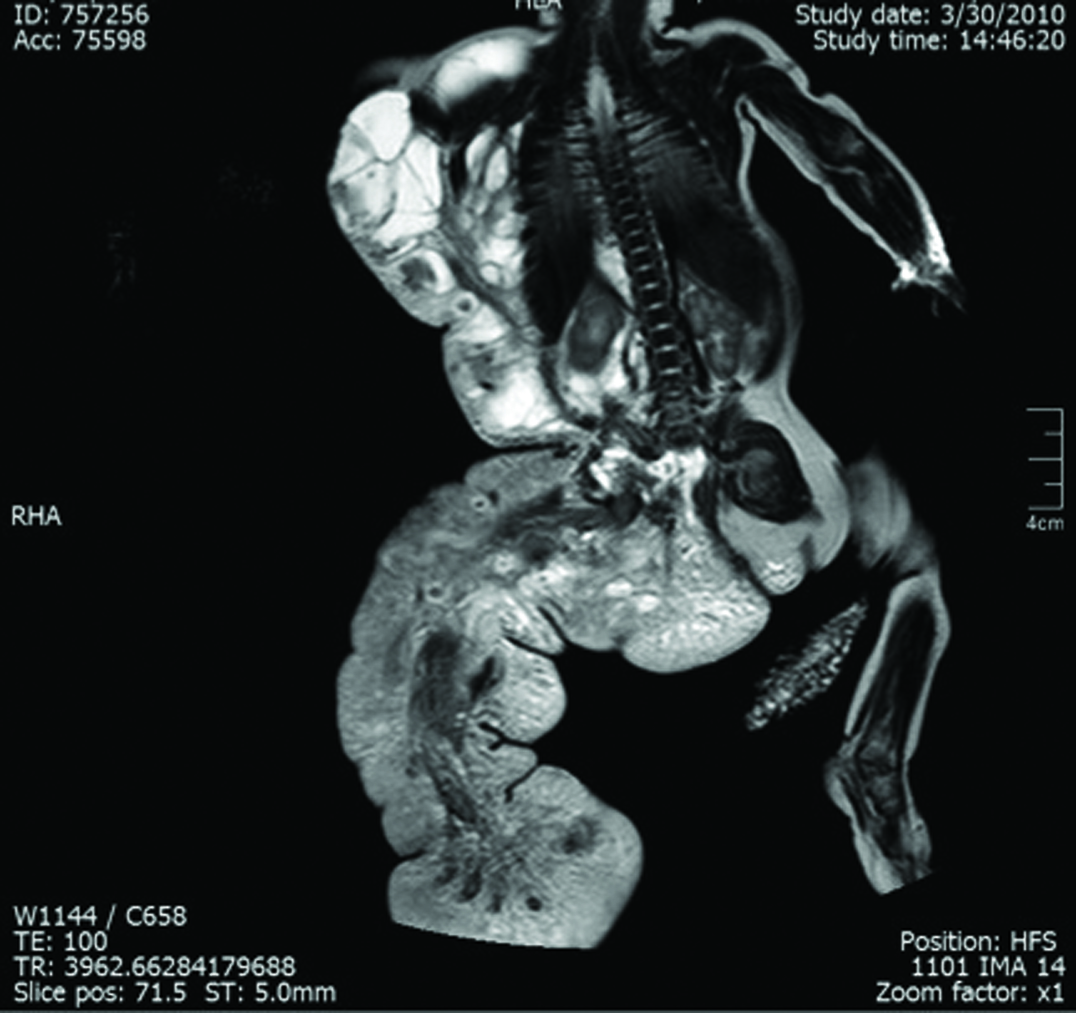
A 4,400 gm girl at birth presented with a giant mass on the right side of her body that extended from the axillary region to half of the trunk and back and was associated with an overgrowth of the lower extremity and absence of toes [Table/Fig-1,2 and 3]. The skin showed several vascular stains on the thigh and flank. Magnetic Resonance Imaging (MRI) showed a combined and extensive vascular malformation with adipose tissue around it, especially in the perineum. The retroperitoneum and mesentery were also involved. Epidermal nevi and scoliosis were not present. The patient had elevated D-dimer and fibrinogen levels. The vascular malformation had two episodes of sudden growth due to internal bleeding observed on ultrasound, requiring several blood transfusions (7 units of red blood cells and 14 units of frozen plasma). Liver, kidney and thyroid function tests were normal. The patient had progressive respiratory distress, requiring mechanical ventilation. The child died from severe Systemic Inflammatory Response Syndrome (SIRS) distress at 54 days of life.
Patient 1: Giant overgrowth on the right side of the body which presented in the axillary region, half of the trunk and back. Vascular anomalies were associated.
Patient 1: Right lower limb with severe overgrowth and deformation of the toes.
Patient 1: Magnetic resonance imaging showing extensive lymphatic and venous malformation and adipose hyperplasia in the perineum.
Case-2
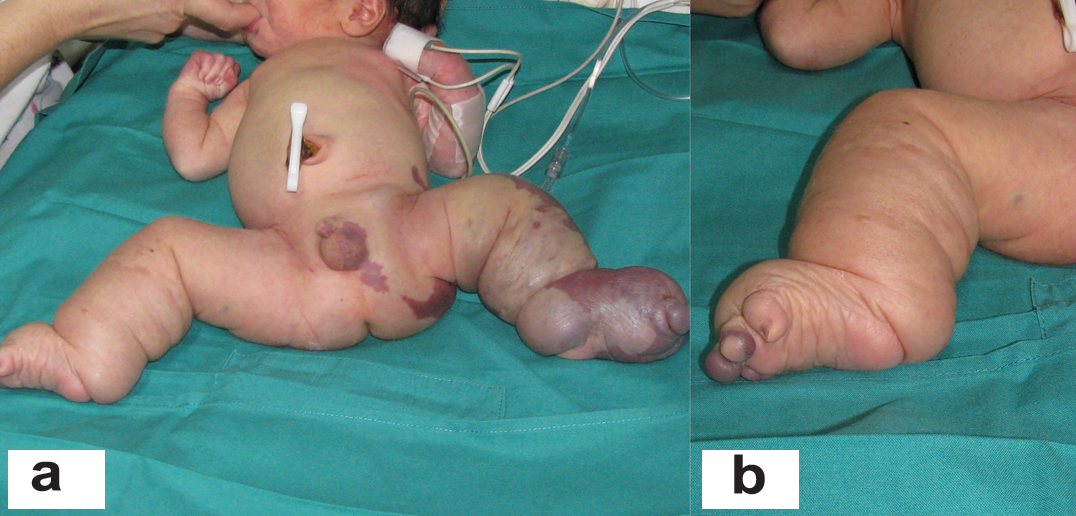
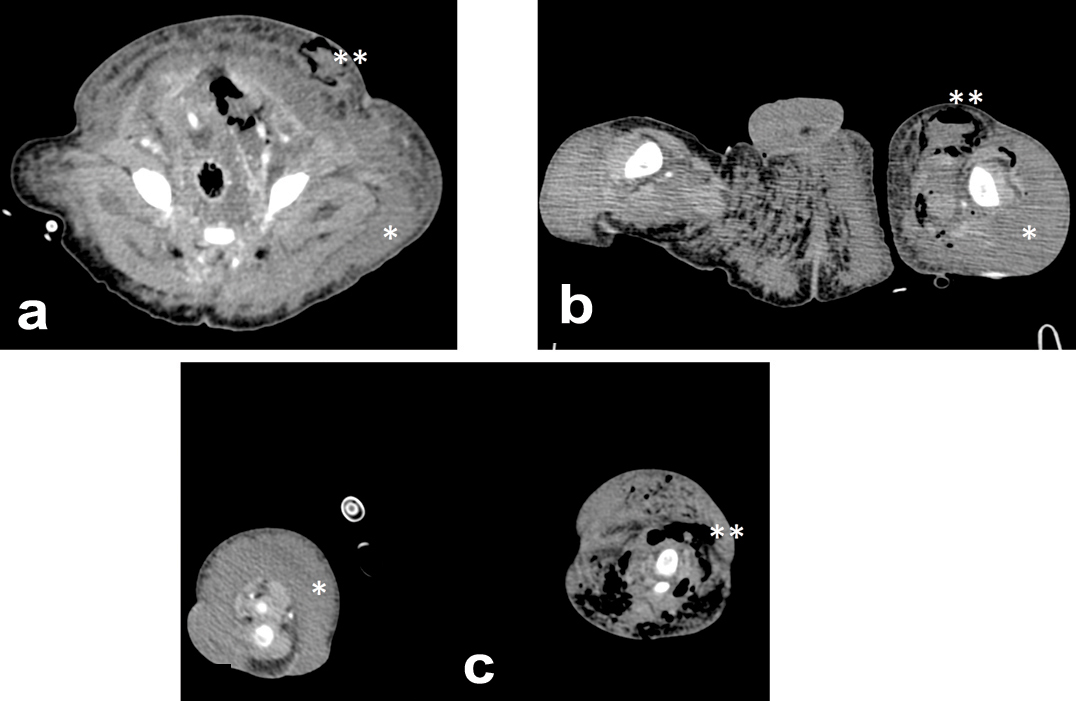
A full-term, 3,808 gm boy presented with overgrowth of the left lower limb and an abnormal foot with no toes (only small buds). On his right lower extremity, he presented with macrodactyly and an increased first web space [Table/Fig-4]. Extensive vascular stains were identified on the left lower extremity and flanks, scrotum and right toes. Angio CT imaging showed extensive retro-peritoneal and subcutaneous lymphatic malformation compromising the abdominal wall, perineum and inguinal region with increased adipose tissue and a giant venous malformation in the thighs and legs [Table/Fig-5]. There were no epidermal nevi and scoliosis present. The coagulation analysis and D-dimer level were normal. At 26 days of age, the infant presented with poor general condition, food refusal, fever, enlargement of both lower extremities, and erythema of the left groin. He required advanced resuscitation and mechanical ventilation due to septic shock of cutaneous origin, with spontaneous drainage of purulent fluid from the lower extremity. The shock was refractory to treatment, and the infant died at 29 days of life. The pathology analysis reported gas gangrene on the left leg and foot inside hamartomatous vascular tissue.
Patient 2: a: Extensive complex, mixed truncal vascular malformations and overgrowth in the left lower limb, perineum and flank with anomalous toes. b: Right foot showing macrodactyly and wide sandal gap. Leg length augmentation.
Patient 2: Extensive lymphatic malformation and increased adipose tissue (*) compromising the abdominal wall a), perineum, inguinal region; b) and legs; c) Gas present due to massive infection (**).
Case-3
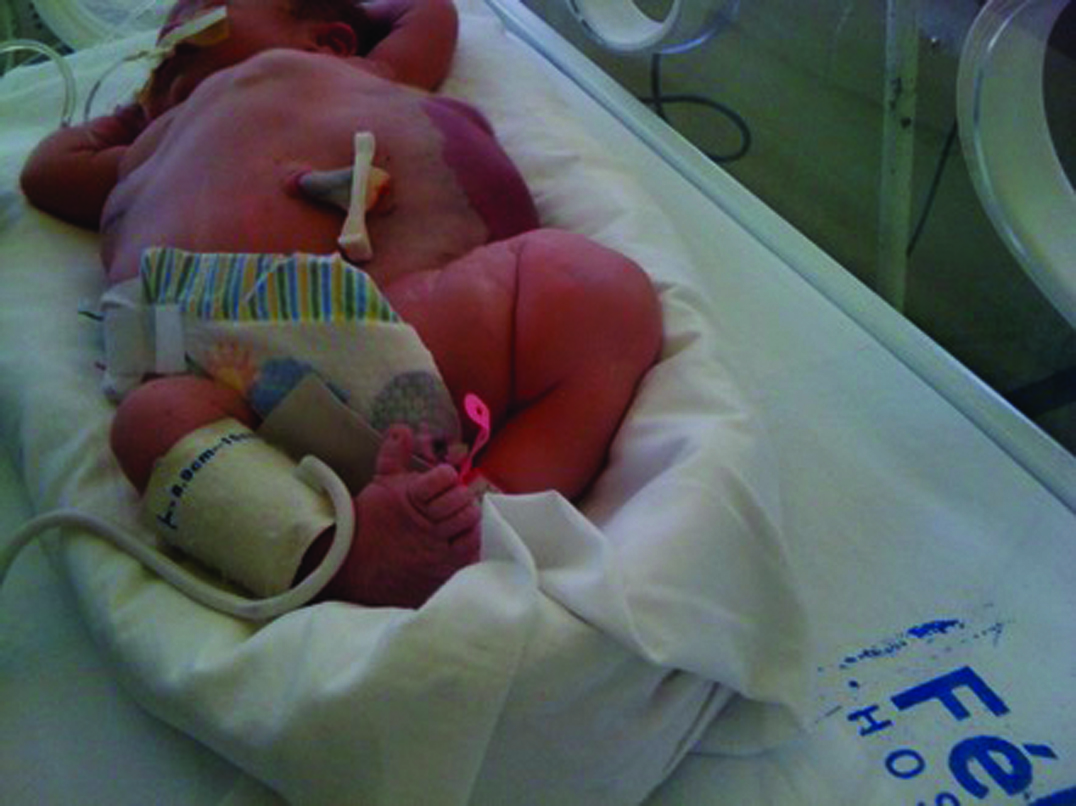
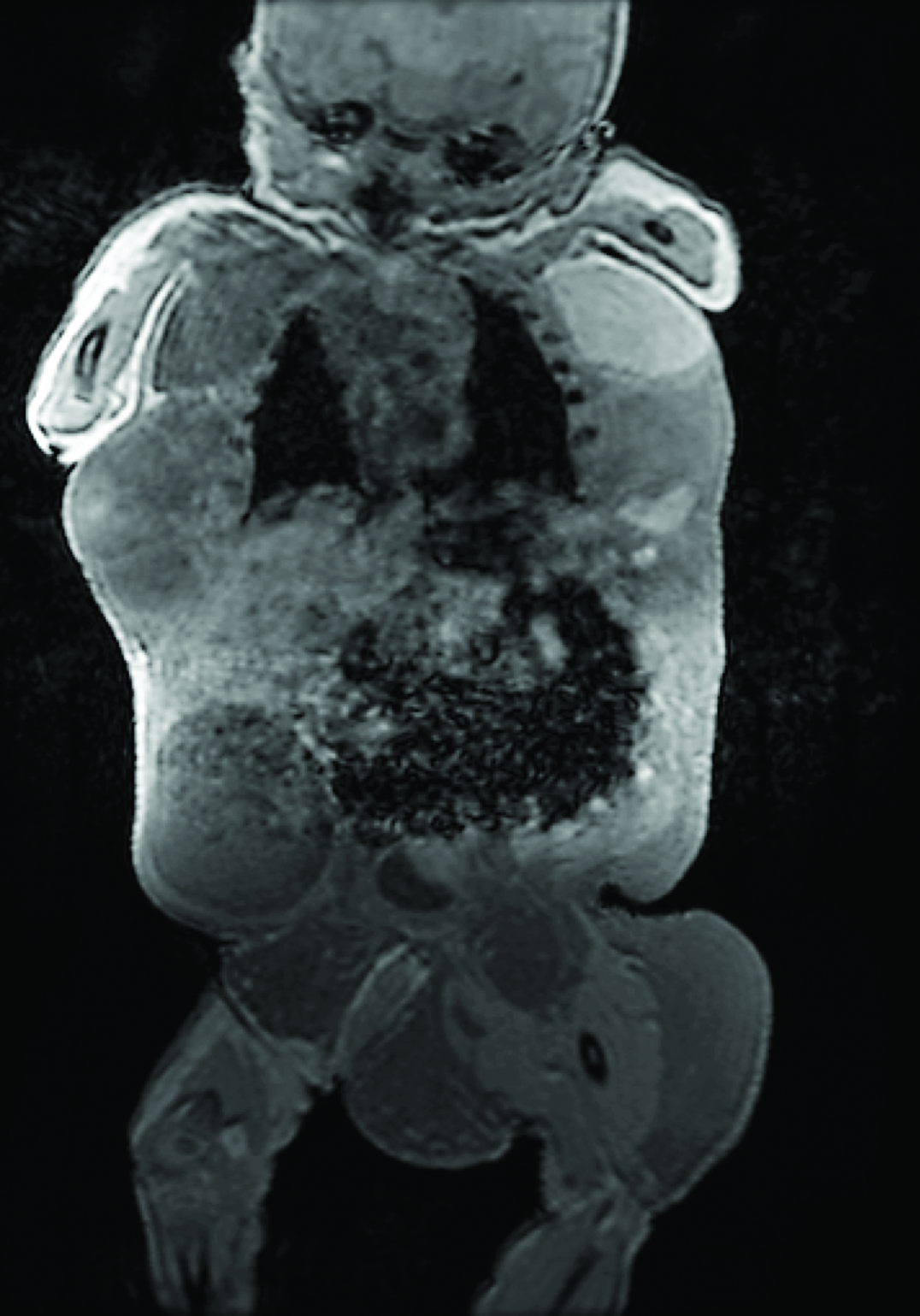
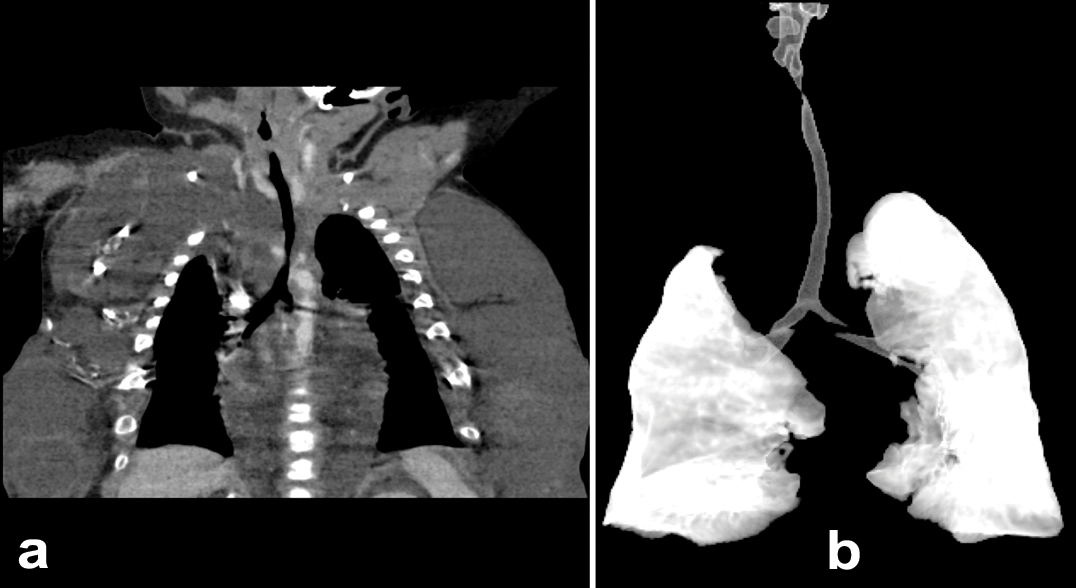
This patient was a full-term, 4,415 gm Caucasian girl in whom multiple tumours in the anterior chest, both flanks, back and left thigh were observed. She also had malformations in the inferior extremities with bilateral club-foot and an increased first web space [Table/Fig-6]. MRI showed an extensive and combined vascular malformation localized in the subcutaneous tissue from the trunk, axillary regions, lateral chest wall, abdomen and pelvis, upper posterior mediastinum and retroperitoneum with adipos ehyperplasia principally in the thoracic wall [Table/Fig-7]. Abdominal ultrasonography showed normal intra-abdominal organs. Epidermal nevi and scoliosis were absent in this patient. She was discharged home in a stable condition at 72 days of life, and injection sclerotherapy was planned after one year. The infant was re-admitted to the hospital at 83 days of life because of apnea episodes during crying and changes of position. CT angiography showed generalized thoracic lymphatic malformation with narrowing of the subglottic trachea [Table/Fig-8]. Both bronchoscopy and laryngoscopy were performed and showed extrinsic compression of the trachea and partial collapse of the left main bronchus. Due to the presence of this large chest vascular malformation that was surrounding the airway, an expectant management was suggested by three paediatric surgeons. She presented with progressive respiratory distress, oxygen requirement, fever and acute anaemia (haemoglobin of 7.5 g/dL). The child died at 106 days of life.
Patient 3: Severe and diffuse truncal overgrowth, right foot with macrodactyly and wide sandal gap. Leg length augmentation.
Patient 3: Magnetic Resonance Imaging (MRI) with extensive and complex combined vascular malformation.
Patient 3: a): Computed tomography angiography with generalized thoracic lymphatic malformation and narrowing of the subglottic trachea; b): Computed tomography reconstruction showing partial collapse of the trachea and the left main bronchus.
Discussion
Sapp et al., in 2007described a common phenotype in previously diagnosed proteus syndrome patients [2]. He characterized it as a “progressive, complex and mixed primarily truncal vascular malformations, de-regulated adipose tissue, varying degrees of scoliosis, and enlarged, yet not severely distorted, bony structures without progressive bony overgrowth”, and he named this entity CLOVE syndrome. Alomari AL expanded this acronym to CLOVES in order to include defects related to the spine, mainly scoliosis, high flow vascular malformations, neural tube defects and tethered cord [1].
It is important to consider that Sapp’s observations were noticed in six patients, whose ages at diagnosis were: 8 weeks, 12, 17, 19, 27 and 55 years old. Therefore, his report lacks description in newborn patients.
A clinical feature in CLOVES syndrome is the truncal lipoma, or para-vertebral trunk fat, that surrounds the paraspinal muscles and the flank [3,4]. However, in the early months or years of life, these areas of deregulated fat tissue may not be easily detectable in the physical exam. In Sapp’s report, like one of our three patients, the 8-week-old patient did not present with this fat tissue behaviour, and neither did the newborn reported by Fernandez-Pineda [5], of whom 2 MRI were taken. A similar description was reported by Harit D [6] and Gucev Z [7]. Sapp described that one of his patients presented with lipomas in late childhood. In Alomari’s report in 2011 [8] from six patients who suffered CLOVES syndrome with complex spinal vascular fast flow lesions, he commented that his first patient presented with a fatty mass at the age of nine months. In further cases, he found heterogeneous fat and lymphatic content instead of a fatty mass. Thus, it can be said that the deregulated fatty tissue is not necessarily an element that must be present in the newborn period with CLOVES syndrome.
All our patients as well as a high percentage of reported cases presented: 1) capillary malformations located in the flank area; 2) lymphatic masses in the chest, sometimes with an intra-abdominal, retroperitoneal and/or pelvic lymphatic involvement; and 3) venous malformations in the lower limbs [2,3]. We believe that these three findings are key to the diagnosis of CLOVES syndrome in the newborn period.
Another skin disorder described is the epidermal nevi, which may be found in the neck, abdomen, flank or limbs. Although, its presence was considered crucial, some cases have been reported without this finding at the newborn stage [1,3,7], and its onset has been observed during growth [9]. In Alomari’s first report comprising 18 patients, 16 of them did not have this anomaly.
The extremities show asymmetric overgrowth, macrodactyly, preferentially affecting the middle digits, sandal gap and wide feet. In two of our patients, the malformations were so severe that there was no recognizable toe normal anatomy. Klein S published an interesting study in which he followed upbone changes in a CLOVES syndrome patient from birth until seven years of age. He mentioned an accelerated, asymmetric skeletal growth with length discrepancy of the lower limbs and pelvic obliquity. Scoliosis was not present at birth, it appeared between two and four years of age [9].
Patients who are born with a combination of capillary, lymphatic and venous malformations have a higher risk of major thrombo embolic events than those who present with less extensive vascular malformations. Alomari AL et al., reviewed angiographic images of a group of 12 patients [10], and they found phlebectasia of the thoracic veins, central veins or both in 11 patients; of those patients, two developed symptomatic pulmonary embolism. We believe that it is important to assess biochemical coagulation elements such as D-dimer and fibrinogen in these patients and to be alert to the need to start appropriate prophylactic measures (e.g., anticoagulation or caval filtration).
Some reports also have described patients presenting with gastrointestinal bleeding associated with CLOVES, probably secondary to vascular malformations in the digestive system [9]. Additionally, Sapp JC and Gucev Z have mentioned central nervous system compromise including hemi-megalencephaly, partial agenesis of the corpus callosum, polymicrogyria and ventriculomegaly [2,7].
Among the disease’s complications, intra-lesional bleeding in the lymphatic vascular malformations is mentioned as well as bacterial infections that could lead to sepsis [3]; both were causes of death in our patients. One of the patients presented with a fulminating sepsis caused by Clostridium perfringens, and the other two had severe growth and bleeding of the vascular malformations.
We concur with Gucev [7] who states “the CLOVE designation should be regarded as a provisional, evolving heuristic at this early stage and that additional clinical data need to be gathered and analysed to refine our understanding of this phenotype.”
We believe that reports of CLOVES syndrome patients may have a potential survival bias. This could occur because patients who survive with CLOVES syndrome are more likely to be reported, leaving out patients who died without this diagnosis because they were not recognized as having CLOVES syndrome. We think that it is very important to recognize this syndrome from birth, bearing in mind that not all clinical findings may be present in the newborn. Epidermal nevi and scoliosis are symptoms that will begin to appear during the second or third year of life. In some cases, adipose proliferation is not clinically evident at the moment of birth. Therefore, these symptoms are not essential in the diagnosis of CLOVES in newborns anymore. It is imperative to know the behaviour of the illness during the growth years.
Conclusion
Based in our series and in the published data, not all of the signs in the acronym are present at birth. Some of them appear with growth.
We propose the following clinical classification:
Classic Presentation: a progressive disease, with clinical features described previously, that appears with child development (epidermal nevi, fatty tissue proliferation, and scoliosis). These children subsequently will grow into adults with variable limitations in everyday life.
Severe neonatal presentation: a disease with the worst spectrum of malformations, with a lower chance of survival, pre-mature death by pulmonary failure secondary to un-manageable intra-thoracic masses, pulmonary embolism, and intra-lesional bleeding or fatal infections.
The previous statement is important because it will affect the information that newborns’ parents will receive as well as the treatment decision-making.
[1]. Alomari AI, CLOVE(S) syndrome: Expanding the acronymAm J Med Genet A 2009 149A(2):294-95. [Google Scholar]
[2]. Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG, Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patientsAm J Med Genet A 2007 143A:2944-58. [Google Scholar]
[3]. Alomari AI, Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndromeClin Dysmorphol 2009 18(1):1-7. [Google Scholar]
[4]. Alomari AI, Comments on the diagnosis and management of cloves syndromePediatr Dermatol 2011 28(2):215-16. [Google Scholar]
[5]. Fernandez-Pineda I, Fajardo M, Chaudry G, Alomari AI, Perinatal clinical and imaging features of CLOVES syndromePediatr Radiol 2010 40(8):1436-39. [Google Scholar]
[6]. Harit D, Clove syndrome with nevus unis lateris: Report of a casePediatr Dermatol 2010 27(3):311-12. [Google Scholar]
[7]. Gucev Z, Tasic V, Jancevska A, Konstantinova MK, Pop-Jordanova N, Trajkovski Z, Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE) syndrome: CNS malformations and seizures may be a component of this disorderAm J Med Genet A 2008 146A(20):2688-90. [Google Scholar]
[8]. Alomari AI, Chaudry G, Rodesch G, Burrows PE, Mulliken JB, Smith ER, Complex spinal-paraspinal fast-flow lesions in CLOVES syndrome: Analysis of clinical and imaging findings in 6 patientsAm J Neuroradiol 2011 32(10):1812-17. [Google Scholar]
[9]. Klein S, Stroberg A, Ghahremani S, Martinez-Agosto JA, Phenotypic progression of skeletal anomalies in CLOVES syndromeAm J Med Genet A 2012 158A(7):1690-95. [Google Scholar]
[10]. Alomari AI, Burrows P, Lee E, Hedequist DJ, Mulliken JB, Fishman SJ, CLOVES syndrome with thoracic and central phlebectasia: Increased risk of pulmonary embolismJ Thorac Cardiovasc Surg 2010 140(2):459-63. [Google Scholar]