Neuromyelitis Optica Spectrum Disorder (NMOSD) is a recently proposed unifying term for Neuromyelitis Optica (NMO), also known as Devic’s disease and related syndromes. It is a relapsing inflammatory demyelinating disease that most commonly affects optic nerves and the spinal cord, leading to sudden vision loss or weakness in one or both eyes, and loss of sensation and bladder function. Though inflammation may also involve the brain, the lesions seen in NMO are different from Multiple Sclerosis (MS) which has a similar clinical course. Attacks of NMOSD tend to be more severe and often different in nature from MS. MS and NMOSD are often confused but require a different course of treatment for optimal results. Here, we describe a patient who had clinical features suggestive of NMO but showed seronegativity for anti-aquaporin antibodies, done twice. Initially he was managed on the lines of MS but showed a deteriorating clinical course on initiation of treatment with interferons. Later, his diagnosis was revised to seronegative NMO and he was started on immunosuppressive therapy with azathioprine to which he showed optimal response and achieved disease stabilization.
Multiple sclerosis, Optic nerves, Spinal cord
Case Report
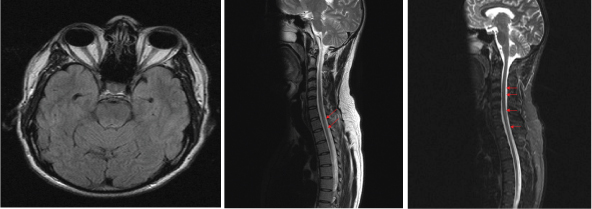
A 36-year-old male patient, presented in Aug 2015, to Neurology OPD at Indian Navy Hospital Ship (INHS) Asvini. He had weakness of both lower limbs along with increased frequency of micturition and painless blurring of left eye vision, of one month duration. There was symmetric progressive weakness in both lower limbs extending from ankle distally to hip region proximally, associated with tightness in both legs while walking and requiring support of one person. He also had erectile dysfunction and reduced sweating in lower half of the body. He had similar episodes of steroid responsive acute onset paraparesis in 2011 and 2012, but without any visual disturbances. Despite adequate steroid therapy, he was left with residual tightness of both legs along with bladder urgency after the second episode. He denied any history to suggest a connective tissue disorder or systemic vasculitis or thrombotic episodes in the past. There was no significant family history. General examination revealed a normotensive and euthymic individual with no rash or lymphadenopathy. Mini Mental State Examination (MMSE) was 30/30 and affect was normal. His cranial nerve examination revealed a visual acuity of 6/24 in left eye with Relative Afferent Pupillary Defect (RAPD) or Marcus Gunn pupil and fundoscopy confirmed papillitis in left fundus. The right eye was normal. Motor examination confirmed a grade 3-4 paraparesis with generalised hyper-reflexia (3+) and Ashworth grade 2 spasticity at both knees. There was a well sustained bilateral ankle clonus. Sensory examination revealed normal spinothalamic and posterior column function. No cerebellar deficits were noted. He had no clinical markers to Vitamin B12/folate deficiency or malabsorption. On investigation his hemogram was normal with an ESR of 12 mm fall at one hour. Liver and renal functions were normal along with normal serum B12 and folate and Angiotensin Converting Enzyme (ACE) levels. His Visual Evoke Potential (VEP) was prolonged in left eye (124 ms) and normal in right eye (102 ms). Detailed autoantibody panel (ANA, RA Factor, Anti ds DNA, ANCA, anti Ro La antibody, anti Jo antibody, anti U1 RNP antibody) was negative. His serology for HIV, Hepatitis B & C were negative. His MRI brain was normal, however, MRI spine was suggestive of diffuse cord oedema and multiple foci of T2 hyperintense cord signal change in the cervico-dorsal region. This signal change was progressive and involving mainly the central cord region, as compared to previous imaging [Table/Fig-1a-c]. His CSF showed mildly elevated proteins (54 mg %) and mild lymphocytic pleocytosis (30 cells/mm3). His CSF oligoclonal bands were negative. His serum anti-aquaporin antibodies were also negative twice from different laboratories (by indirect immunofluorescence). He was initially managed as a primary CNS demyelinating disease (Optico-spinal variant of MS) with pulsed steroids followed by interferon Beta 1a (Inj. Avonex 30mcg IM/week) therapy, with partial clinical improvement in paraparesis (Grade 4) and left eye vision (6/12) over 2-3 months. However, he continued to have multiple spinal relapses (3 in one year) despite regular compliance to interferon therapy. Subsequently, his diagnosis was then revised to seronegative NMOSD and he was started on immunosuppressive therapy with Azathioprine (3 mg/kg/day) to which he showed good response and achieved disease stabilization. He is planned for Rituximab therapy in case of fresh disease activity.
(a) Normal brain MRI (Axial FLAIR Image); (b) Initial MRI spine (Oct 11): diffuse cord oedema and signal change D5-D11; (c) Recent MRI spine (Aug 2015): aggravation in lesions with extensive myelitis involving most segments of cervical and dorsal cord from C3 to D12.
Discussion
Neuromyelitis Optica (NMO) is an autoimmune, inflammatory disease of the central nervous system that usually affects the optic nerves and spinal cord. Nearly a century ago in 1894, Eugene Devic and his student Fernand Gault documented optic nerve and spinal cord involvement in a series of 16 patients with features of MS and hence, NMO is commonly known as “Devic’s Disease” [1]. Since the discovery of Aquaporin-4 antibody (AQP4-Immunoglobulin G [IgG]), a specific serum marker in 2004, major advancements are made in the diagnosis of NMO [2]. The 2006 revised diagnostic criteria for neuromyelitis optica incorporated AQP4-IgG serology [3]. In 2007, the term NMOSD was newly introduced. NMOSD included patients with limited form of NMO with AQP4-IgG seropositivity apart from patients who have typical NMO presentation [4]. Also, NMOSD included AQP4-IgG seropositive patients with autoimmune disorders and patients diagnosed with opticospinal MS which was previously considered as MS phenotype [5]. However, seronegative patients (AQP4-IgG negative) with clinical features of NMOSD can be encountered mainly because of the variable sensitivity of the technique involved in detection of AQP4-IgG antibody [6,7].
NMOSD Diagnostic Criteria
The international panel for NMO diagnosis which convened 07 times between Oct 2011 to Nov 2013 subsumed NMO into a single term NMOSD, which is a broader terminology to include patients with neither optic neuritis nor myelitis but which occur eventually in almost all patients [8].
Criteria were developed for both NMOSD with AQP4-IgG and NMOSD without AQP4-IgG. NMOSD diagnostic criteria are illustrated in [Table/Fig-2] [9-15]. A diagnosis of NMOSD is made if at least one out of six core clinical characteristics are met and AQP4-IgG is detected. The six core clinical characteristics denote the six classic regions affected in central nervous system, namely, optic nerve, spinal cord, area postrema of the dorsal medulla, brain stem, diencephalon and cerebrum [16-20]. Also, NMOSD has certain characteristic clinical presentations which include bilateral optic neuritis involving optic chiasma causing field defect [14], complete spinal cord syndrome with tonic spasms [14] and area postrema syndrome causing intractable hiccups, nausea or vomiting [15]. However, no characteristic presentation is pathognomonic for NMOSD.
NMOSD diagnostic criteria [9-15].
| Core clinical characteristics | Optic neuritis Acute myelitis Area postrema syndrome: unexplained hiccups or nausea and vomiting Acute brainstem syndrome Symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions Symptomatic cerebral syndrome with NMOSD-typical brain lesions
|
| Diagnostic criteria for NMOSD with AQP4-IgG | At least one core clinical characteristic Positive test for AQP4-IgG (cell-based assay strongly recommended) Exclusion of alternative diagnoses
|
| Diagnostic criteria for NMOSD without AQP4-IgG or NMOSD with unknown AQP4-IgG status | At least two core clinical characteristics and meeting all of the following requirements:
At least one core clinical characteristic must be optic neuritis, acute myelitis with LETM, or area postrema syndrome Dissemination in space (2 or more different core clinical characteristics) Fulfillment of additional MRI requirements
Negative tests for AQP4-IgG (cell-based assay strongly recommended), or testing unavailable Exclusion of alternative diagnoses
|
AQP4 = aquaporin-4; IgG = immunoglobulin G; LETM = longitudinally extensive transverse myelitis lesions; NMOSD = neuromyelitis optica spectrum disorders
The consensus made stringent diagnostic requirement for patients who are seronegative (AQP4-IgG negative) or for whom testing could not be done. Such individuals should have experienced two or more core clinical characteristics and other supportive MRI features. Of the two or more different core clinical events, atleast one must be either optic neuritis, transverse myelitis or an area postrema syndrome – the three most common clinical characteristics of NMOSD [21].
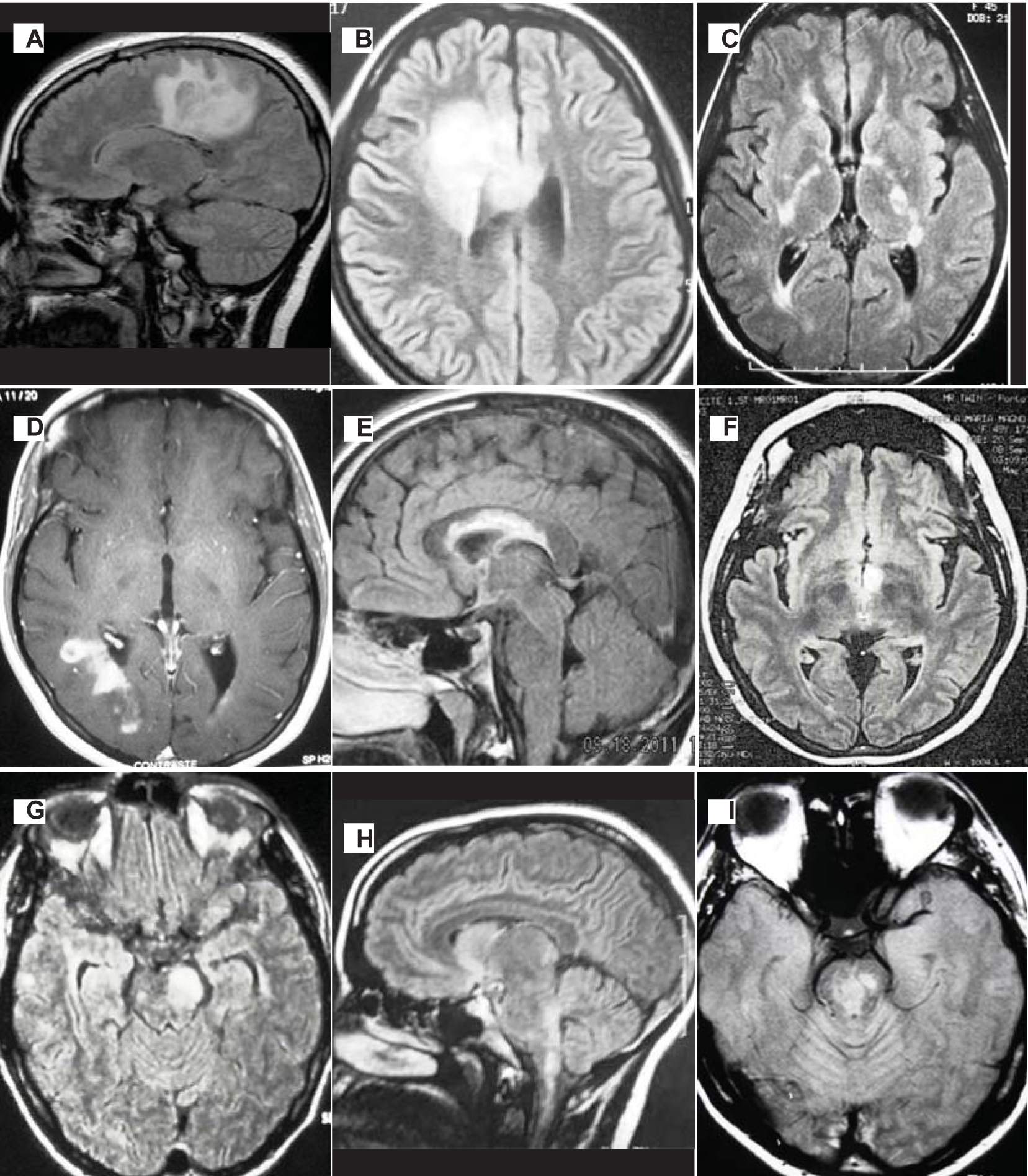
Several characteristic patterns involving brain, spinal cord and optic nerve are highly suggestive of NMOSD. The occurrence of Longitudinally Extensive Transverse Myelitis (LETM) which involves 03 or more vertebral segments with acute myelitis is very uncommon in multiple sclerosis. Such spinal cord involvement in neuroimaging is most suggestive of NMOSD whereas Multiple sclerosis cord lesions are usually 01 vertebral segment long [22]. Even the extensive cord involvement is not pathognomonic of NMOSD as 7% to 14% of initial myelitis in seropositive NMOSD patients do not meet the radiologic LETM definition [23]. This makes NMOSD a differential diagnosis even for short myelitis lesions. About 7% of patients have NMOSD typical patterns in brain MRI, though not pathognomonic for NMOSD [23]. The typical brain lesions seen in NMOSD are depicted in [Table/Fig-3].
Typical brain lesions in NMOSD (a) Extensive edematous white matter lesion; (b) Tumefactive frontal lobe lesion (c) Bilateral lesions in the posterior limb of the internal capsule; (d) Cloud-like gadolinium-enhanced lesion; (e) Heterogeneous extensive callosal lesion; (f) Bilateral lesions in the hypothalamus; (g) Large mesencephalic lesion; (h) Lesion involving area postrema; (i) Central brainstem lesion.
The expanded spectrum of neuromyelitis optica: evidences for a new definition. Arq Neuropsiquiatr. 2012;70(10):807-813) (Adapted with permission from Marco A Lana-Peixoto.)
AQP4-IgG Serologic Testing and Anti-MOG Antibodies
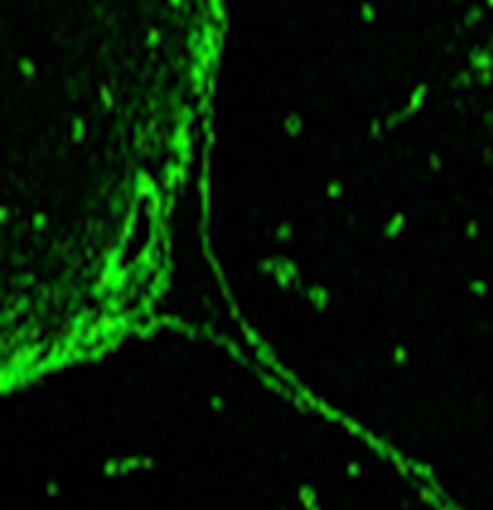
Technological advancements in AQP4-IgG assays have greatly improved the sensitivity in diagnosing NMOSD. Various techniques commonly used are indirect immunofluorescence [Table/Fig-4], ELISA and cell based assays which include microscopy or flow cytometry-based detection. Cell based assays are to be used whenever possible because they carry a mean sensitivity of 76.7% in pooled analysis as they employ antibody optimization [24]. Indirect immunofluorescence assays and ELISA have lower sensitivity (mean sensitivity 63% to 64% each). False positive results with non-cell based assays are also high (0.5% to 1.3% compared to 0.1% in cell based assays) [24].
Representative picture showing Aquaporin-4 antibodies in indirect immunofluorescence.
The practical difficulty is that cell based assays are not widely available even in the tertiary care settings, even when they have the best sensitivity of 75-80%. The commonly available testing methods by IIF or ELISA can be false negative in 30% to 35% cases resulting in diagnostic challenges in many cases.
Patients who fulfill the diagnostic criteria for NMOSD but are seronegative for AQP4-IgG (usually when assessed by non-cell based assays) represent a diagnostic challenge [25]. A minority of such patients have been reported to have serum Myelin Oligodendrocyte Glycoprotein (MOG) antibodies at detectable levels [26]. NMOSD patients showing positive anti-MOG antibodies and AQP4-IgG negative are usually younger at age, less frequently female and are less likely to relapse [25,26]. The baseline clinical differences hint towards a possibility that Anti-MOG NMOSD patients might have a different underlying pathogenesis and a milder disease as compared to classic Anti-AQP4 disease.
Our patient had 02 core clinical characteristics in the form of optic neuritis and acute myelitis occurring as a result of two clinical attacks in 2011 and 2013. His serum AQP4-IgG tested by indirect immunofluorescence (the available detection method) done twice was negative. Also, there was no alternate diagnosis to explain the disease process along with normal MRI Brain. Hence, he satisfied all criteria required for the diagnosis of NMOSD without AQP4-IgG i.e., seronegative NMOSD.
Conclusion
This case highlights the importance of understanding the expanding spectrum of NMOSD, and correct application of the latest revised diagnostic criteria (2013), along with the limitation of conventional NMO antibody testing by IIF and ELISA. Thus, whenever clinical suspicion is high for a NMOSD and conventional AQP4-IgG tests are negative, an effort should be made to obtain a cell based assay with higher sensitivity and to further check on anti-MOG antibody status. This is important to plan appropriate therapy and improve patient outcomes in NMOSD. Treatment modalities for attack prevention in NMO and MS differ significantly and certain immunotherapies for MS can aggravate NMO, as was seen in this case.
[1]. Jarius Swildemann B, The history of neuromyelitis opticaJournal of Neuroinflammation 2013 10(1):8 [Google Scholar]
[2]. Lennon V, Wingerchuk D, Kryzer T, Pittock S, Lucchinetti C, Fujihara K, A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosisThe Lancet 2004 364(9451):2106-12. [Google Scholar]
[3]. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG, Revised diagnostic criteria for neuromyelitis opticaNeurology 2006 66:1485-89. [Google Scholar]
[4]. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG, The spectrum of neuromyelitis opticaLancet Neurol 2007 6:805-15. [Google Scholar]
[5]. Kuroiwa Y, Shibasaki H, Epidemiologic and clinical studies of multiple sclerosis in JapanNeurology 1976 26:8-10. [Google Scholar]
[6]. Kimbrough D, Fujihara K, Jacob A, Lana-Peixoto M, Isabel Leite M, Levy M, Treatment of neuromyelitis optica: Review and recommendationsMultiple Sclerosis and Related Disorders 2012 1(4):180-87. [Google Scholar]
[7]. Min JH, Kim BJ, Lee KH, Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorderMult Scler 2012 18:113-15. [Google Scholar]
[8]. Nagaishi A, Takagi M, Umemura A, Tanaka M, Kitagawa Y, Matsui M, Clinical features of neuromyelitis optica in a large Japanese cohort: Comparison between phenotypesJournal of Neurology, Neurosurgery & Psychiatry 2011 82(12):1360-64. [Google Scholar]
[9]. Matiello M, Lennon V, Jacob A, Pittock S, Lucchinetti C, Wingerchuk D, NMO-IgG predicts the outcome of recurrent optic neuritisNeurology 2008 70(23):2197-2200. [Google Scholar]
[10]. Weinshenker B, Wingerchuk D, Vukusic S, Linbo L, Pittock S, Lucchinetti C, Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitisAnnals of Neurology 2006 59(3):566-69. [Google Scholar]
[11]. Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, Brainstem manifestations in neuromyelitis optica: A multicenter study of 258 patientsMult Scler 2013 20(7):843-47. [Google Scholar]
[12]. Poppe A, Lapierre Y, Melançon D, Lowden D, Wardell L, Fullerton L, Neuromyelitis optica with hypothalamic involvementMult Scler 2005 11(5):617-21. [Google Scholar]
[13]. Kim W, Kim SH, Lee SH, Li XF, Kim HJ, Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorderMult Scler 2011 17:1107-12. [Google Scholar]
[14]. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG, The clinical course of neuromyelitis optica (Devic’s syndrome)Neurology 1999 53:1107-14. [Google Scholar]
[15]. Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y, Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis opticaNeurology 2005 65:1479-82. [Google Scholar]
[16]. Bichuetti D, Rivero R, Oliveira D, Souza N, Abdala N, Oliveira E, Neuromyelitis optica: Brain abnormalities in a Brazilian cohortArq Neuro-Psiquiatr 2008 66(1):1-4. [Google Scholar]
[17]. Nakamura M, Misu T, Fujihara K, Miyazawa I, Nakashima I, Takahashi T, Occurrence of acute large and edematous callosal lesions in neuromyelitis opticaMult Scler 2009 15(6):695-700. [Google Scholar]
[18]. Matsushita T, Isobe N, Matsuoka T, Ishizu T, Kawano Y, Yoshiura T, Extensive vasogenic edema of anti-aquaporin-4 antibody-related brain lesionsMult Scler 2009 15(9):1113-17. [Google Scholar]
[19]. Saiki S, Ueno Y, Moritani T, Sato T, Sekine T, Kawajiri S, Extensive hemispheric lesions with radiological evidence of blood–brain barrier integrity in a patient with neuromyelitis opticaJournal of the Neurological Sciences 2009 284(1-2):217-19. [Google Scholar]
[20]. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA, Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expressionArch Neurol 2006 63:964-68. [Google Scholar]
[21]. Apiwattanakul M, Popescu B, Matiello M, Weinshenker B, Lucchinetti C, Lennon V, Intractable vomiting as the initial presentation of neuromyelitis opticaAnnals of Neurology 2010 68(5):757-61. [Google Scholar]
[22]. Huh S, Min J, Kim W, Kim S, Kim H, Kim B, The usefulness of brain MRI at onset in the differentiation of multiple sclerosis and seropositive neuromyelitis optica spectrum disordersMultiple Sclerosis Journal 2013 20(6):695-704. [Google Scholar]
[23]. Flanagan E, Weinshenker B, Krecke K, Lennon V, Lucchinetti C, McKeon A, Short myelitis lesions in aquaporin-4-IgG–positive neuromyelitis optica spectrum disordersJAMA Neurology 2015 72(1):81 [Google Scholar]
[24]. Kitley J, Woodhall M, Waters P, Leite M, Devenney E, Craig J, Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotypeNeurology 2012 79(12):1273-77. [Google Scholar]
[25]. Kitley J, Waters P, Woodhall M, Leite M, Murchison A, George J, Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodiesJAMA Neurology 2014 71(3):276 [Google Scholar]
[26]. Mao Z, Lu Z, Hu X, Sato D, Callegaro D, Lana-Peixoto M, Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disordersNeurology 2014 83(12):1122-23. [Google Scholar]