Bardet Biedl Syndrome – A Report of Two Cases with Otolaryngologic Symptoms
Mahendra K Singh1, Shrinkhal2, Sidharth Pradhan3, Priyanko Chakraborty4
1 Professor and Head, Department of Ophthalmology, Institute of Medical Sciences, BHU, Varanasi, Uttar Pradesh, India.
2 Junior Resident, Department of Ophthalmology, Institute of Medical Sciences, BHU, Varanasi, Uttar Pradesh, India.
3 Junior Resident, Department of Otorhinolaryngology, Institute of Medical Sciences, BHU, Varanasi, Uttar Pradesh, India.
4 Junior Resident, Department of Otorhinolaryngology, Institute of Medical Sciences, BHU, Varanasi, Uttar Pradesh, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sidharth Pradhan, Room no.130, Sushruta Hostel, Trauma Centre Campus, IMS BHU, Varanasi, Uttar Pradesh, India.
E-mail: pradhansidharth.1987@gmail.com
Bardet-Biedl Syndrome (BBS) is a rare autosomal recessive disorder characterized primarily by rod-cone dystrophy, postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction. We present two cases of this syndrome, both female, who presented with complaints of nyctalopia and mental retardation, and additionally one of them had sensorineural hearing loss while the other had serous otitis media. Hearing loss being a rare presentation is worth reporting. Both the patients were given a course of vitamin A and the parents were counseled regarding the prognosis and additional complications associated with the syndrome.
Hearing loss, Postaxial polydactyly, Rod-cone dystrophy, Serous otitis media
Case Report
We hereby report two cases of BBS who presented to the Department of Ophthalmology with slightly variable presentation.
Case-1
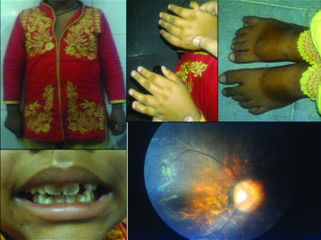
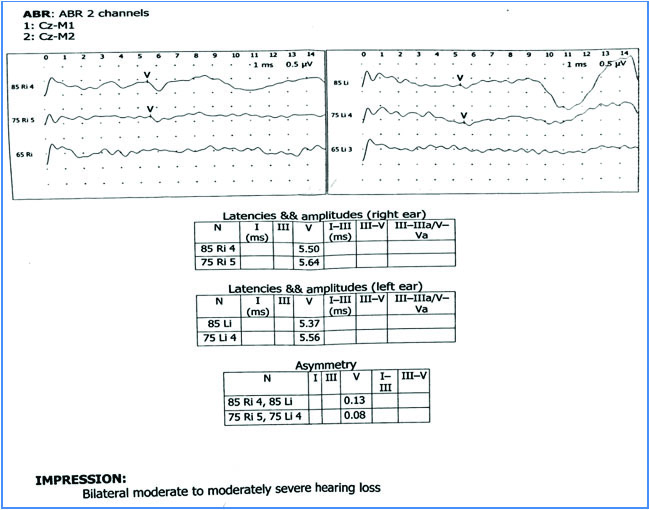
An eight-year-old girl presented with the chief complaint of diminution of vision in both eyes at night since one year (as told by father). Diminution of night vision was progressive in nature. She had history of operation in her feet due to some congenital deformity. Although enrolled in school, her IQ was much lower than others of the same age group. She had diminished hearing. Her visual acuity at presentation was 6/12 in both eyes on Snellen’s chart. There was no familial history of similar symptoms and her parents were not consanguineous. Systemic examination revealed truncal obesity to be present, with polydactyly and brachydactyly involving both hands and feet. Dental malocclusion was also present [Table/Fig-1]. On ocular slit lamp examination, anterior segment was normal. Fundus examination by indirect ophthalmoscope revealed bilateral diffuse retinal dystrophy, arteriolar attenuation, disc pallor and absence of any pigments [Table/Fig-1]. Electroretinogram showed depressed rod function. Bilateral tympanic membrane was intact on otoscopic examination. Tuning fork test pointed towards bilateral sensori-neural hearing loss which was confirmed by Brainstem Evoked Response Audiometry (BERA). BERA revealed that an identifiable wave V was obtained at 75 dBnHL in both ears with latencies of 5.64 ms in right ear and 5.56 ms in left ear at 75 dBnHL, thus confirming bilateral moderate to moderately severe sensori-neural hearing loss to be present [Table/Fig-2]. Her social quotient was much lower than her peers. Hypogonadism was not evident on USG of abdomen. There was no evidence of paraparesis or muscle weakness. The diagnosis of BBS with retinitis pigmentosa sine pigmento was made from the above findings. The parents were explained about the nature of the disease, poor visual prognosis and a course of vitamin A (15000 IU of Retinol palmitate) was prescribed for a duration of 30 days. The patient was then referred to the Department of Cardiology and Nephrology to exclude the associated anomalies but the parents refused further evaluation and didn’t come for follow up.
Case no.1 showing (clockwise from top left) central obesity, polydactyly and brachydactyly involving hand and feet, fundus finding, and dental malocclusion.
Case-2
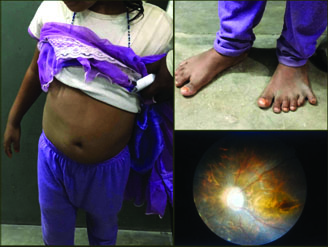
A seven-year-old female child presented with similar complaints of decreased vision at night since one year, which was progressive along with polydactyly and brachydactyly involving hand and left foot. She had been operated in her left hand for the same [Table/Fig-3]. Additionally she had sero-sanguinous ear discharge for last 15 days and was congenitally mute. Visual acuity was 6/9 in both eyes on Snellen’s chart. Examination of the fundus revealed diffuse retinal dystrophy without pigmentation. Otoscopic examination showed serosanguinous discharge which on mopping revealed Grade 3 retraction of pars tensa in both ears with bubbles behind tympanic membrane in left ear. Audiometry showed mild conductive hearing loss. On per-oral examination, both tonsils were enlarged (Grade 3). Her social quotient was very low. Hypogonadism was again not evident on USG of abdomen. A course of 15000 IU of vitamin A was given for a month along with antihistaminics for serous otitis media. Tympanotomy with placement of Grommet and adenotonsillectomy were advised to the patient’s parents, but they refused any further surgical procedures. The patient was referred to the Department of Nephrology and Cardiology for further assessment but was lost to follow up.
Case no. 2 showing (clockwise from left) central obesity, polydactyly and brachydactyly involving feet, and fundus finding.
Discussion
BBS is an autosomal recessive condition with multisystem involvement. Primarily characterized by rod-cone dystrophy (sometimes called atypical retinitis pigmentosa), postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction [1], it also includes additional symptoms such as hepatic fibrosis, diabetes mellitus, reproductive abnormalities, endocrinological disturbances, short stature, developmental delay, and speech deficits [1]. BBS is distinguished from the much rarer Laurence-Moon syndrome, in which retinal pigmentary degeneration, mental retardation, and hypogonadism occur in conjunction with progressive spastic paraparesis and distal muscle weakness, but without polydactyly [1,2]. In both of the above cases, rod-cone dystrophy, central obesity, mental retardation and polydactyly were present along with speech disabilities. While one of the patients had sensori-neural hearing loss, the other had serous otitis media with Grade 3 tonsillitis and mild conductive hearing loss. Neither of the patients had hypogonadism.
BBS is genetically heterogeneous, with four loci mapped to date which includes BBS1 (11q13) – commonest, BBS2 (16q22), BBS3 (3p13) and BBS4 (15q21) [3–6]. Retinal dystrophy is the most important feature of the disorder. Attenuated vessels, mottling, granularity of the Retinal Pigment Epithelium (RPE) and waxy pale appearance of the disc are typical fundus findings in Retinitis Pigmentosa (RP) associated with this syndrome (atypical RP). Truncal obesity is second major feature. Limb abnormalities are present in majority of cases, polydactyly being the commonest. Brachydactyly and rarely syndactyly may also be found. Syndactyly if present is usually partial and confined to the feet. Hypogonadism is a usual finding in male patients, but is much less common in females. Renal abnormalities, dental anomalies and mental retardation are present in majority of patients [7]. Rarer presentations include behavioral abnormalities, hearing loss (conductive and rarely sensorineural), neurological and motor defects, asthma, heart defects and diabetes mellitus [1].
BBS is a chronic disease without any specific cure. Parents require proper counseling with proper advice regarding the life threatening systemic complications of the syndrome. A course of vitamin A may be given for nyctalopia. Speech therapy and proper rehabilitation may be necessary for some children affected by this syndrome.
Conclusion
Hearing loss is a rare manifestation of BBS. Hypogonadism isn’t a frequent feature in females affected by BBS as was found in the above two cases. A patient of BBS must be followed up regularly for early identification of any of the life modifying or threatening complications and appropriate management of the same.
[1]. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA, New criteria for improved diagnosis of Bardet-Biedl Syndrome: results of a population surveyJ Med Genet 1999 36:437-46. [Google Scholar]
[2]. Laurence JZ, Moon RC, Four cases of “retinitis pigmentosa” occurring in the same family, and accompanied by general imperfections of developmentOphthalmol Rev 1866 2:32-41. [Google Scholar]
[3]. Leppert M, Baird L, Anderson KL, Otterud B, Lupski JJ, Lewis RA, Bardet-Biedl syndrome is linked to DNA markers on chromosome 11q and is genetically heterogeneousNat Genet 1994 7:108-12. [Google Scholar]
[4]. Kwitek-Black AE, Carmi R, Duyk GM, Buetow KH, Elbedour K, Parvari R, Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneityNat Genet 1993 5:392-96. [Google Scholar]
[5]. Sheffield VC, Carmi R, Kwitek-Black A, Rokhlina T, Nishimura D, Duyk GM, Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mappingHum Mol Genet 1994 3:1331-35. [Google Scholar]
[6]. Carmi R, Rokhlina T, Kwitek-Black AE, Elbedour K, Nishimura D, Stone EM, Use of a DNA pooling strategy to identify a human obesity syndrome locus on chromosome 15Hum Mol Genet 1995 4:9-13. [Google Scholar]
[7]. Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndromeN Engl J Med 1989 321:1002-09. [Google Scholar]