Choroid plexus hypertrophy, Nevus flaemmus, Port wine stain, Tramtrack calcification
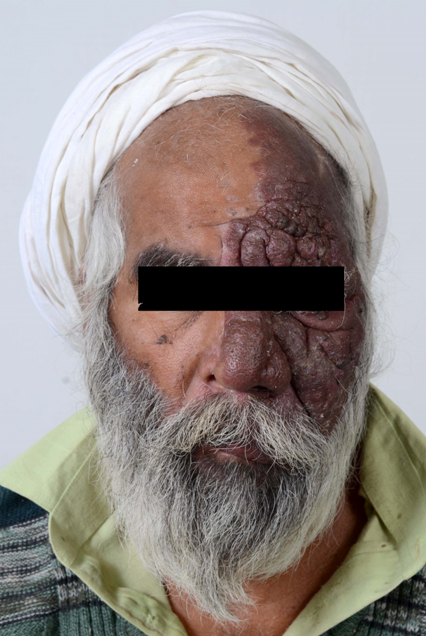
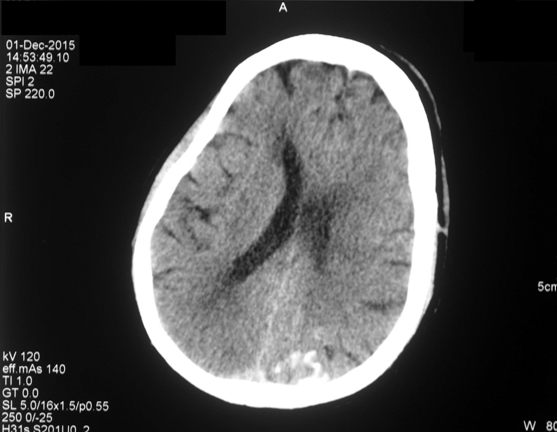
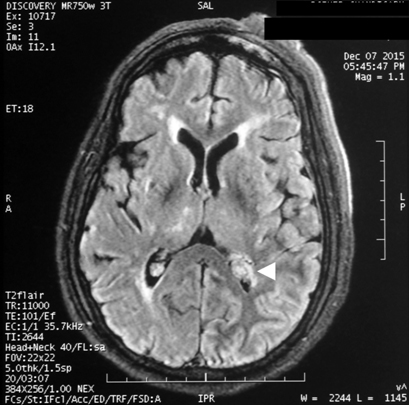
A 55-year-old gentleman presented to the emergency wing with generalized tonic-clonic seizures and altered sensorium. He was on carbamazepine for focal seizures since 20 years; however, the compliance was poor. He also had gradually progressive visual loss since last 20 years; however, there was no devlepomental delay or mental retardation. On examination, he was in post-ictal state (Glasgow Coma score 9/12). A large elevated, purple, hemangiomatous lesion was noted over left half of the face in the distribution of the ophthalmic and maxillary divisions of the trigeminal nerve [Table/Fig-1]. The hematological parameters, serum electrolytes, calcium, magnesium and plasma glucose were within normal range. A non contrast Computed Tomography (CT) of the brain revealed intracerebral calcification involving the left occipital cortex [Table/Fig-2]. Contrast enhanced Magnetic Resonance Imaging (MRI) of brain suggested peri-ventricular white matter changed and enlargement of the choroid plexus [Table/Fig-3]. In view of the constellation of findings, a diagnosis of Sturge- Weber Syndrome was made. He was treated with 20 mg/kg of phenytoin followed by maintenance doses. Sensorium improved over the next 12 hours. On recovery, he was discharged on oral anti-epileptics and advised to follow up with neurology and ophthalmology services for management of seizures and glaucoma.
A 55-year-old male patient with a large progressive hemangiomatous lesion (portwine stain) over the left half of the face in V1 and V2 distribution.
V1- ophthalmic and V2- maxillary
A non contrast CT brain showing intracerebral calcification involving the left occipital cortex;
Contrast enhanced MRI brain (FLAIR sequence) demonstrating the enlarged choroid plexus (arrow head) in a 55-year-old male patient with Sturge-Weber Syndrome.
Sturge-Weber Syndrome is a sporadic, congenital neurocutaneous disorder, characterized by intracranial leptomeningeal vascular malformations associated with a facial portwine stain (nevus flaemmus).
The portwine stain is usually found along the distribution of the ophthalmic (V1) and maxillary (V2) divisions of the trigeminal nerve and may also involve the scalp, neck, trunk or limbs. It may remain static in extent but can undergo progressive hypertrophy, darken and become nodular in upto 65% of the patients by the fifth decade [1]. About 8% of the individuals with a portwine stain may have an underlying Sturge-Weber Syndrome and the association is upto 78% if the lesion involves the entire V1 distribution [2].
The leptomeningeal angiomas cause vascular steal and cortical ischemia leading to the cerebral atrophy and/or dystrophic calcifications [3]. These are best seen on MRI, while CT is the best modality to demonstrate the ‘tramtrack calcification’. Neurological manifestations include recurrent, refractory seizures (focal or generalized), transient neurological deficit, developmental delay, mental retardation and visual impairment (most often due to glaucoma).
Glaucoma can affect 30-70% of patients and ipsilateral eye is mostly affected [4]. Other ocular abnormalities could be choroidal, conjunctival hemangiomas and heterochromia of the iris.
Treatment is symptomatic with antiepileptics, antiglaucoma drugs and laser therapy for portwine stain. Low dose Aspirin has been studied in the prevention of stroke like episodes and seizures [5]. Surgical intervention is reserved for patients with refractory seizures and uncontrolled glaucoma.
In conclusion, all patients with portwine lesions over the face, especially involving V1 region of trigeminal nerve, should be evaluated for Sturge-Weber Syndrome, as early diagnosis and prompt treatment may reduce the incidence of neurologic sequelae, and may prevent irreversible blindness.
[1]. Geronemus RG, Ashinoff R, The medical necessity of evaluation and treatment of port-wine stainsJ Dermatol Surg Oncol 1991 17:76-79. [Google Scholar]
[2]. Ch’ng S, Tan ST, Facial portwine stains - clinical stratification and risks of neuro-ocular involvementJ Plast Reconstr Aesthet Surg 2008 61:889-93. [Google Scholar]
[3]. Aylett SE, Neville BG, Cross JH, Boyd S, Chong WK, Kirkham FJ, Sturge-Weber syndrome: Cerebral haemodynamics during seizure activityDev Med Child Neurol 1999 41:480-85. [Google Scholar]
[4]. Sujansky E, Conradi S, Outcome of Sturge-Weber syndrome in 52 adultsAm J Med Genet 1995 57:35-45. [Google Scholar]
[5]. Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM, Survey of aspirin use in Sturge-Weber syndromeJ Child Neurol 2011 26:692-702. [Google Scholar]