Renal Primitive Neuroectodermal Tumour: Case Report of a Rare Entity
Sai Chandana Gali1, B.N. Kumarguru2, Balachandra Bhat3, A.S. Ramaswamy4, M. Udaya Kumar5
1 Tutor and Postgraduate, Department of Pathology, PESIMSR, Kuppam, Andhra Pradesh, India.
2 Assistant Professor, Department of Pathology, PESIMSR, Kuppam, Andhra Pradesh, India.
3 Consultant Pathologist, Department of Pathology, PESIMSR, Kuppam, Andhra Pradesh, India.
4 Former Professor and Head, Department of Pathology, PESIMSR, Kuppam, Andhra Pradesh, India.
5 Professor and Head, Department of Pathology, PESIMSR, Kuppam, Andhra Pradesh, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sai Chandana Gali, Tutor and Postgraduate, Department of Pathology, PESIMSR, Kuppam- 517425, Andhra Pradesh, India.
E-mail: chandana.gali@gmail.com
The peripheral Primitive Neuroectodermal Tumour (PNET) is a member of the family of small round cell tumours. PNET is more aggressive in kidney when compared to the other sites. It usually presents in childhood or adolescence. It has an aggressive clinical course and may process towards metastatic disease culminating in death. A 24-year-old female presented with left sided abdominal swelling. Abdominal ultrasound confirmed a heterogeneous left renal mass. Consequently the patient underwent nephrectomy of left kidney and left oophorectomy. Grossly, the tumour involved almost entire kidney, showed multi-lobular, grey, glistening appearance with focal haemorrhagic areas. Histologically, the tumour cells were arranged in diffuse infiltrating sheets, cohesive lobules, Homer-Wright rosettes and perivascular pseudo-rosettes. Individual tumour cells were small round cells with scant cytoplasm and round nuclei having dispersed chromatin. Features were suggestive of PNET. Immunohistochemistry showed tumour cells displaying strong membrane positivity for MIC 2. Renal PNET needs to be differentiated from other primary and metastatic renal round-cell tumours. Most of the cases of renal PNET have poor response to standard treatment of combined surgical resection, post-operative irradiation, and chemotherapy. PNET is a rare primary tumour in the kidney. Histopathological diagnosis has to be confirmed by immunophenotyping of the tumour cells.
Histopathology, Immunohistochemistry, Kidney, Nephrectomy
Case Report
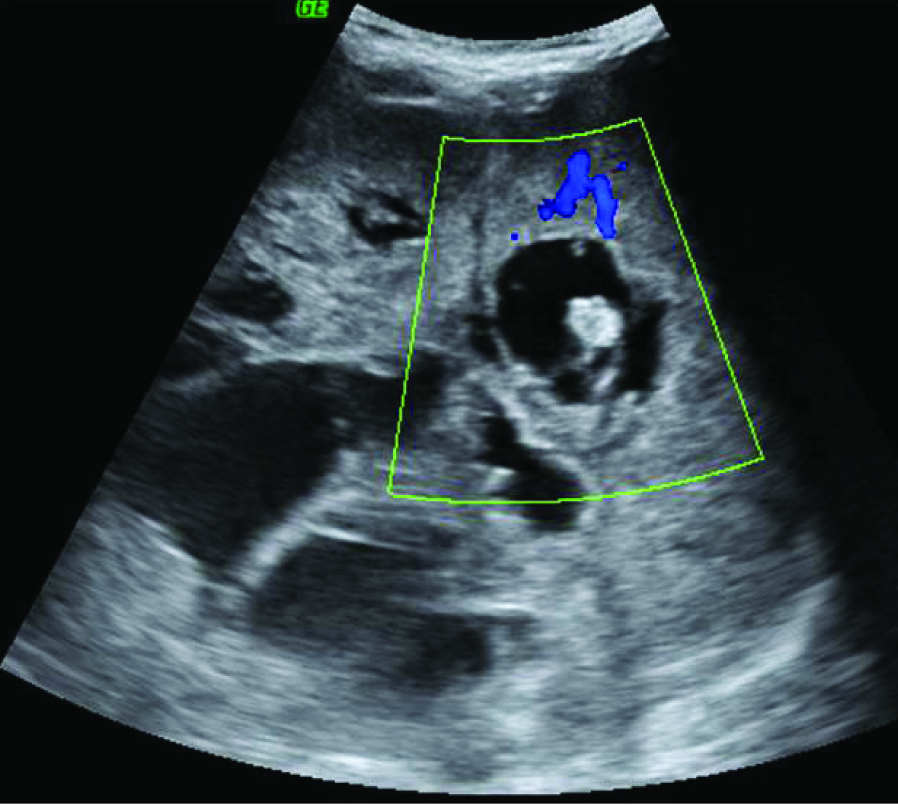
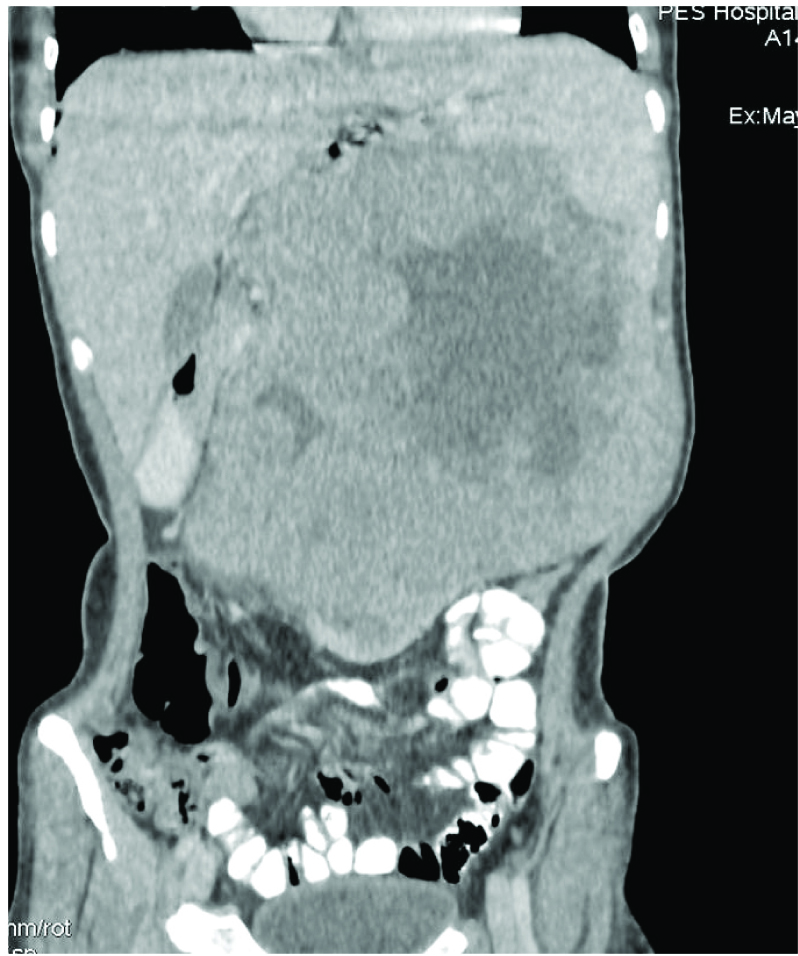
A 24-year-old female patient presented with left sided abdominal swelling of two months duration. It was associated with history of significant weight loss. On physical examination, abdominal distension was noticed in left hypochondrial and umbilical region. The mass was firm in consistency. Abdominal ultrasonography showed heterogeneous mass arising from left kidney [Table/Fig-1] Contrast Enhanced Computerized Tomography (CECT) of abdomen showed large heterogeneous mass replacing almost entire left kidney, sparing a small segment in the lower pole [Table/Fig-2]. The mass was crossing midline and causing external mass effect on vascular tributaries. Right kidney and other abdominal organs were normal. Routine investigations showed microcytic hypochromic anemia, haemoglobin being 8.5gm/dl. Urine analysis showed protenuria and haematuria. Provisional clinical diagnosis of left renal cell carcinoma was offered. Patient underwent left radical nephrectomy. Bilateral polycystic ovaries were detected. Hence, left oophorectomy and right ovary drilling was performed.
Abdominal ultrasound showing a heterogeneous left renal mass;
Contrast Enhanced Computerized Tomography (CECT) of abdomen showing large heterogeneous mass.
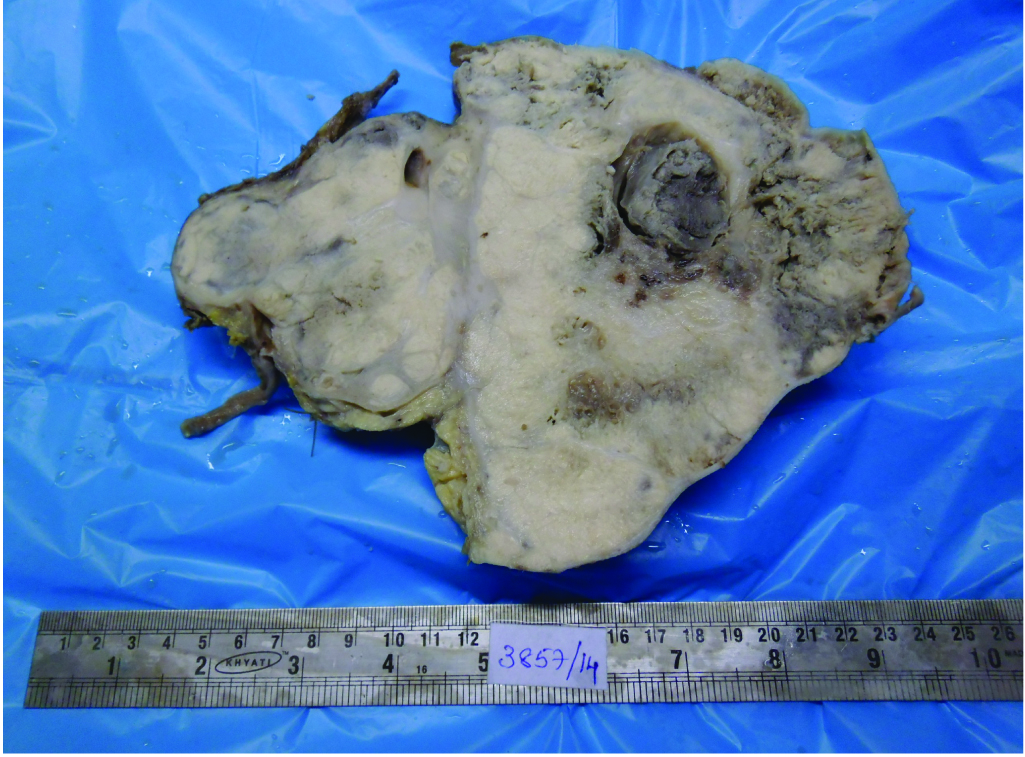
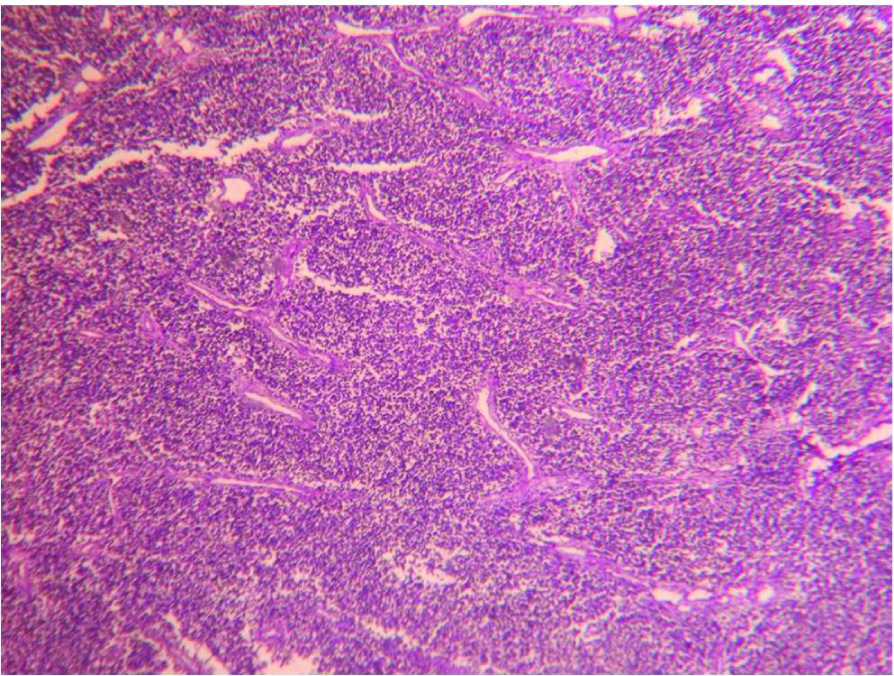
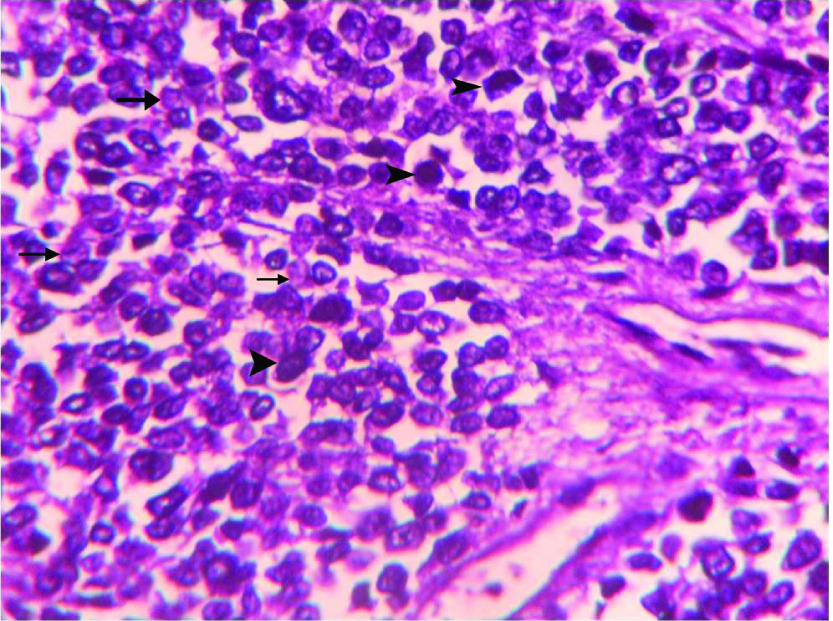
We received left radical nephrectomy specimen measuring 24x22x14 cm and weighing 900 gm. External surface was bosselated with loss of original shape. Ureter measured 9 cm in length. Renal vessels were not identified. Renal pelvis appeared to be involved. Cut-section showed a tumour involving almost entire kidney. The tumour had multi-lobular, grey, glistening appearance with focal haemorrhagic areas [Table/Fig-3]. Also, seen were grey-yellow areas with necrotic and small cystic areas. On Microscopy, tumour cells were arranged in infiltrating sheets [Table/Fig-4], cohesive lobules, Homer-Wright rosettes [Table/Fig-5a] and perivascular pseudo-rosettes [Table/Fig-5b]. Individual tumour cells were small round cells with scant cytoplasm, high nuclear cytoplasmic ratio and round nuclei having dispersed chromatin. Also, seen were interspersed small dark cells [Table/Fig-6]. Tumour cells showed variable chromatin distribution pattern from hyperchromatic to vesicular nuclei with inconspicuous nucleoli. Prominent vasculature and areas of necrosis were seen. Adjacent renal parenchyma was edematous. Tumour cells infiltrated pelvicalyceal area, renal capsule and Gerota fascia. Involvement of renal sinus and renal vessels could not be made out. Ureteric cut margin was not involved. Left adrenal gland was free of tumour. A diagnosis of small round cell tumour suggesting renal Primitive Neuroectodermal Tumour (PNET) was offered. Immunohistochemistry (IHC) was performed on tumour tissue. Neoplastic cells showed strong membranous positivity for MIC2 [Table/Fig-7] and were negative for Wilm tumor 1 (WT1), Myogenin, Leukocyte Common Antigen (LCA), Cytokeratin (CK) Synaptophysin and Chromogranin. A final diagnosis of renal PNET was made on morphological and immunophenotypic grounds.
Cut-section of left renal mass showing multi-lobular grey glistening tumour tissue.
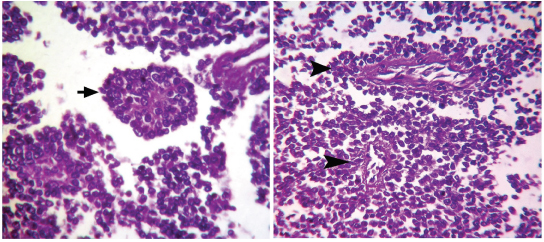
Microphotograph of tissue section of left kidney showing infiltrating sheets of small round cells. (H&E,100X)
(a) Microphotograph of tumour tissue showing Homer-Wright rosette (arrow). (H&E, 400X); (b) Microphotograph of tumour tissue showing perivascular pseudo rosettes (arrow heads). (H&E, 400X).
Microphotograph of tumour tissue showing small round cells having scanty cytoplasm and nuclei showing variable chromatin pattern {light (arrows) and dark nuclei (arrow heads)}. (H&E,400X).
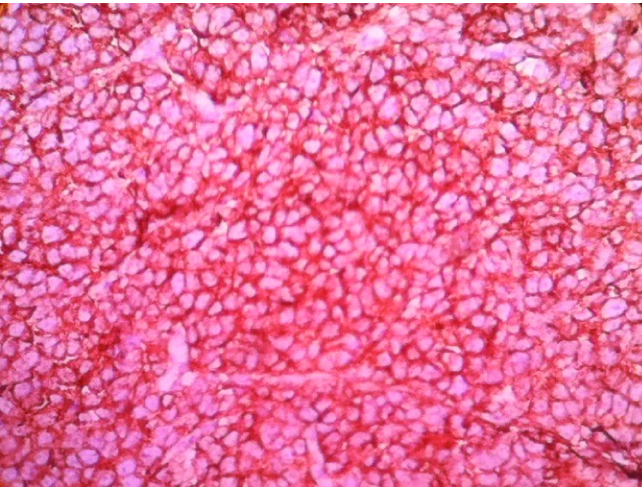
Microphotograph of tumour tissue showing strong membrane positivity of neoplastic cells for MIC-2. (IHC, 400X).
Left ovary measured 4.5x2.5x2 cm. External surface showed multiple cysts. Cut-section showed multiple cysts largest measuring 1cm in diameter. Microscopy showed multiple cystic follicles and was uninvolved by tumour.
Follow up ultrasonography revealed mild ascites and no pelvic lymphadenopathy. Post-operative period was uneventful and patient was discharged in stable condition.
Discussion
Peripheral PNET was first recognized by Arthur Purdy Stout in the year 1918. PNET belongs to family of “small round cell tumours” [1]. It is derived from primitive cells of neural crest. It commonly involves bone and soft tissues in adolescent patients. Small round cell tumours of the kidney are a heterogeneous group of neoplasms which have overlapping morphological features and different prognostic and therapeutic implications [2]. Renal PNET is a rare entity and is more aggressive than in other sites [1]. Most frequently renal PNET arises from medullary region or pelvic region of kidney [3]. It usually presents in childhood or adolescence. It has an aggressive clinical course and may process towards metastatic disease culminating in death [1].
PNET is an extraordinarily rare primary tumour in the kidney [4]. Renal PNET was first reported by Seemayer et al., Mean age of presentation being 27 years [5], the tumour has a predilection for males. Abdominal pain of recent (weeks) or sudden onset, flank pain and gross haematuria are the most common presenting symptoms. Fever, weight loss and bone pain are other less frequent manifestations. A palpable abdominal or flank mass was detected in less than 25% of cases [6]. In the present case, a 24-year-old female patient presented with left sided abdominal swelling of two months duration. A heterogeneous left renal mass was detected on abdominal ultrasonography and CECT scan.
Grossly, tumour had multi-lobular, grey, glistening appearance with focal haemorrhagic, cystic and necrotic areas. Pomara G et al., Erdogan S et al., Krozak T et al., observed similar gross morphological features [1,2,5]. Histologic hallmark of PNET is formation of Homer-Wright rosettes or pseudo-rosettes involving hyperchromatic cells [5]. Both the types of rosettes were observed in the present case.
Renal PNET/Ewing tumour should be differentiated from other small blue cell tumours arising in kidney such as blastemal predominant Wilms tumour, neuroblastoma, rhabdomyosarcoma, lymphoblastic lymphoma, small cell neuroendocrine carcinoma, desmoplastic small blue cell tumour and synovial sarcoma [4]. IHC and molecular studies have a key role in establishing an accurate diagnosis and in differentiating from other small round cell tumours [7].
Immunohistochemical recognition of MIC2 gene product (CD 99) is a highly sensitive marker for Ewing/PNET family of tumours [8]. In the present case, neoplastic cells showed strong membranous positivity for MIC2 and negative for battery of other markers. Immunophenotype favored the diagnosis of renal PNET.
Several theories have been proposed for explanation of genesis of PNET arising at peripheral sites. One of them is presence of aberrant neural crest cell. Another explanation is origin from neural ramification of celiac plexus that innervates the kidney [2]. Less popular hypothesis includes one which considers PNET to be derived from pleuripotent germ cell and other favours a mesenchymal stem cell origin. Peripheral type may be found in the genitourinary tract, testis, ovary, uterus, or pancreas [9].
Cytogenetic studies can also be used to demonstrate the association of PNET/ Ewing sarcoma with translocation of long arms of chromosome 11 and 22, t (11;22) (q22;q12) [10]. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) detection of specific fusion transcripts is recognized as a very powerful tool in the diagnosis of Ewing/PNET family of tumours [9]. Most common metastatic sites were lymph nodes, lung, liver and bone [2].
Surgery, chemotherapy and radiotherapy are the modalities of treatment followed for the management of renal PNET. Current standard chemotherapy includes doxorubicin, vincristine, and cyclophosphamide alternated with ifosfamide and etoposide [7] with 5-year survival rate of 45-55% [1].
Conclusion
Renal PNET is a rare primary neoplasm of kidney. Histological diagnosis is challenging as it mimics other small round cell tumours. Diligent search should be made to look for the presence of rosettes and pseudo rosettes, as these features provide important diagnostic clues. Cellular immunophenotype helps to differentiate it from other small round cell tumours having similar microscopic features. MIC-2 positivity helps to confirm the histological diagnosis.
[1]. Pomara G, Cappello F, Cuttano MG, Rappa F, Morelli G, Mancini P, Primitive neuroectodermal tumour (PNET) of the kidney: a case reportBMC Cancer 2004 4:3 [Google Scholar]
[2]. Erdogan S, Ersoz C, Gonlusen G, Tuganalp A, Primitive neuroectodermal tumour of the kidney: A case report and review of literatureAPJ 2005 2:49-53. [Google Scholar]
[3]. Ordonez NG, Rosai J, Urinary tract : Kidney, renal pelvis, and ureter; BladderIn: Rosai and Ackerman’s Surgical Pathology 2011 10th EdPhiladelphiaMosby Elsevier:1102-246.Rosai J, editors [Google Scholar]
[4]. Castro EC, Parwani AV, Ewing sarcoma/primitive neuroectodermal tumour of the kidney: Two unusual presentations of a rare tumourCase Rep Med 2012 :1-7. [Google Scholar]
[5]. Kroczak T, Sharda R, Drachenberg D, Essawi T, Renal primitive neuroectodermal tumour: Case series and brief reviewCan Urol Assoc J 2014 8:241-44. [Google Scholar]
[6]. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, Pathology and Genetics of Tumours of the urinary system and male genital organsWorld Health Organization Classification of Tumours 2004 LyonIARC Press:83-4. [Google Scholar]
[7]. Shringarpure SS, Venkatraman M, Raja T, Mani R, A case of primitive neuroectodermal tumour of the kidney and review of the literatureIndian J Pathol Microbiol 2012 55:587-88. [Google Scholar]
[8]. Karpate A, Menon S, Basak R, Yuvaraja TB, Tongaonkar HB, Desai SB, Ewing sarcoma/primitive neuroectodermal tumour of the kidney: clinicopathologic analysis of 34 casesAnn Diagn Pathol 2012 16:267-74. [Google Scholar]
[9]. Su JR, Yang S, Lo KY, Primitive neuroectodermal tumour (PNET) of the kidneyJ Urol Roc 2001 12(4):180-82. [Google Scholar]
[10]. Kakkar S, Gupta D, Kaur G, Rana V, Primary primitive neuroectodermal tumour of kidney: A rare case report with diagnostic challengeIndian J Pathol Microbiol 2014 57:298-300. [Google Scholar]