Primary Ewing Sarcoma of Sphenoid Bone with Intracranial Extension: A Common Tumour at an Uncommon Location
Guddi Rani Singh1, Vijayanand Choudhary2, Rawi Agrawal3
1 Senior Resident, Department of Pathology, Indira Gandhi Institute of Medical Sciences, Patna, Bihar, India.
2 Additional Professor, Department of Pathology, Indira Gandhi Institute of Medical Sciences, Patna, Bihar, India.
3 Senior Resident, Department of Pathology, Indira Gandhi Institute of Medical Sciences, Patna, Bihar, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Rawi Agrawal, 305, Ushakunj Apartment, Ambedkar Path, Jagdeopath, Bailey Road-800014, Patna, Bihar, India.
E-mail: agrawalrawi@gmail.com
Primary Ewing Sarcoma of the cranial bone is rare, contributing to only 1% of all Ewing Sarcomas. Primary cranial Ewing Sarcoma occurs most commonly in temporal bone followed by parietal and occipital bones. Sphenoid bone is less commonly involved. We report a case of Ewing Sarcoma of the sphenoid bone with intra-cranial extension in a 20-month-old boy. On CT scan a provisional diagnosis of rhabdomyosarcoma was made. Fine Needle Aspiration Cytology (FNAC) and histopathological examination of core needle biopsy showed small round cell tumour. On Immunohistochemistry (IHC), CD99 (MIC2) and FLI 1 were strongly positive and final diagnosis of Ewing Sarcoma was made. Considering the rarity of this unusual site, we report a case of primary Ewing Sarcoma arising in the sphenoid bone with erosion of adjacent bones and intra-cranial extension.
CD99 (MIC2), Cranial tumour, Primitive neuroectodermal tumor, Small round cell tumour
Case Report
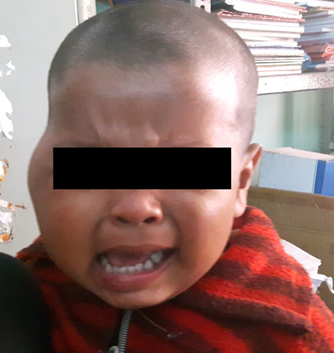
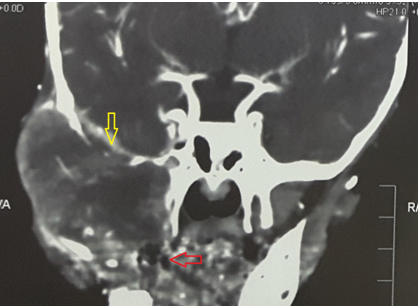
A 20-month-old male child presented in the paediatric surgery OPD with chief complaints of a rapidly increasing, painful swelling over the right cheek for over 2 months [Table/Fig-1]. There was no history of trauma. On physical examination, there was gross facial asymmetry with an ill-defined swelling extending from the lateral canthus of right eye up to the right cheek. The right palpebral fissure was decreased in size and movement of the right eye was restricted, but visual acuity was apparently normal. There were no neurological deficits. Haematological examinations were within normal limits. CT scan showed a 5cmx6cm enhancing soft tissue mass arising in the body and right wing of sphenoid bone with intra-cranial extension [Table/Fig-2]. Right cavernous sinus, right maxillary sinus and right retro-orbital space were involved by the tumour and erosion of the frontal bone, right temporal bone, right zygomatic bone, lateral wall of the right orbit and the right mandible were seen. Provisional diagnosis of rhabdomyosarcoma was made on CT scan.
Patient with right cheek swelling;
CT scan showing mass arising in the body and right wing of sphenoid (red arrow) bone extending intracranially (yellow arrow).
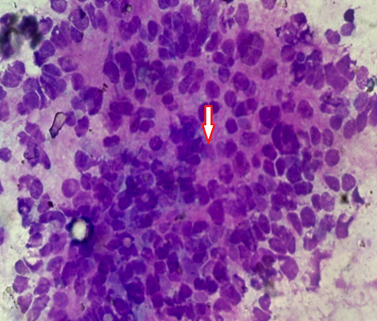
FNAC from the cheek swelling was done. The smears were cellular and showed loose clusters and dispersed sheets of small round to oval cells with high nucleus:cytoplasmic (N:C) ratio. Rosette formation was also seen [Table/Fig-3].
FNAC smear showing small round to oval cells with high N/C ratio with rosette (arrow) formation. (Giemsa stain, × 400);
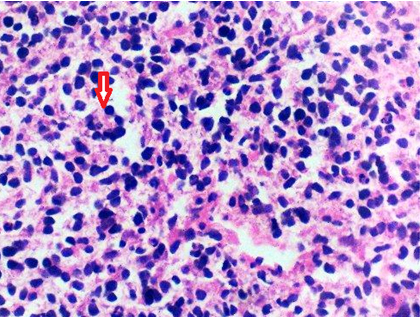
Core needle biopsy from the tumour was taken and sent for histopathological examination. Haematoxylin and Eosin (H&E) stained sections showed a tumour composed of small round cells. Tumour cells had a high N/C ratio, round to oval uniform nuclei with finely dispersed chromatin, inconspicuous nucleoli and scant cytoplasm. Cytoplasm was vacuolated at places. Rosette formation was seen [Table/Fig-4]. On Periodic Acid–Schiff (PAS) staining glycogen was not detected in the cytoplasm of cells.
Microsection showing small round cell tumour with rosette (arrow) formation. (H&E stain, × 400).
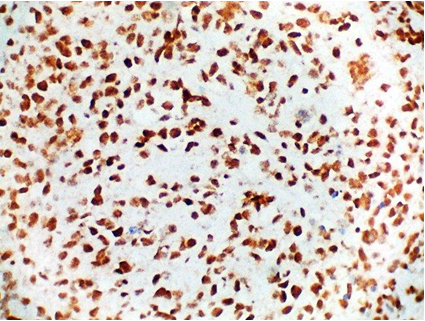
On immunohistochemistry, CD99 (MIC2) and FLI 1 were positive. CD 56 was focally positive. CD 45, S-100, Synaptophysin and Neuron Specific Enolase (NSE) were negative. Ki-67 labelling was 70% [Table/Fig-5,6 and 7]. A final diagnosis of Ewing Sarcoma was made.
Tumour cells showing CD 99 positivity (IHC stain, × 400);
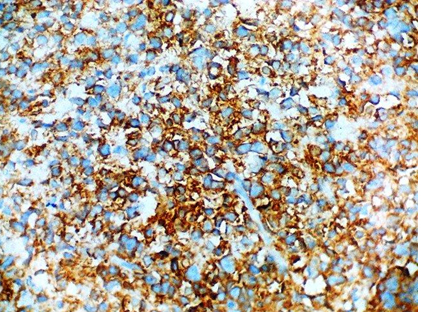
Tumour cells showing FLI 1 positivity. (IHC stain, × 400).
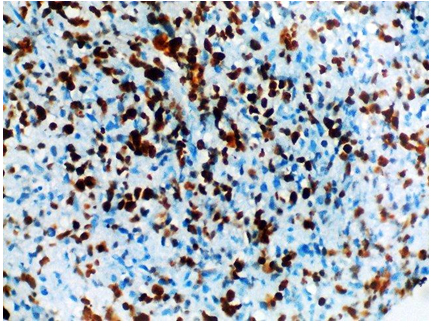
Tumour cells showing 70% Ki-67 labeling. (IHC stain, × 400).
As the tumour extended intra-cranially complete excision was not possible and debulking surgery was done. This was followed by radiotherapy and chemotherapy. Chemotherapy with Vincristine, Doxorubicin and Cyclophosphamisde, alternating with Ifosfamide and Etoposide was given initially for 12 weeks. Radiation was given at the local site. Thereafter, treatment was completed with the same chemotherapeutic agents was planned for another 24 weeks. But the child succumbed to sepsis at 16th week.
Discussion
Ewing Sarcoma is an aggressive malignant small round cell tumour usually seen in children and young adults with a peak incidence in the second decade of life [1]. It is the second most common paediatric bone tumour [2]. There is no sex difference in incidence of Ewing sarcoma. Primary Ewing Sarcoma of skull is very rare and its incidence is less than 1% [3]. Most common bone involved by ewing sarcoma in skull is temporal bone followed by parietal bone and occipital bone. Sphenoid and ethmoid bones are least commonly involved [4]. It most commonly presents as a painful swelling that enlarges rapidly over a period of few weeks to few months (as in our case also) [5]. In case of intra-cranial extension with or without involvement of neural structures, patient may present with features of increased intra-cranial pressure and focal neurological deficit [6]. But in our case inspite of intra-cranial extension, patient did not have any focal neurological deficit. The most common CT finding is isodense lesion with marked heterogenous enhancement. Biopsy and immunohistochemistry is essential for definitive diagnosis.
Ewing Sarcoma is part of a large family of round-blue-cell tumours. It is composed of sheets of small cells with high N/C ratio. Cytoplasm of individual cells is usually scanty and eosinophilic with glycogen vacuoles. Cytoplasmic glycogen is important in differential diagnosis, but it may be lost during formalin fixation and paraffin embedding and hence, not demonstrable by Periodic acid–Schiff (PAS stain), as in our case [7]. Nuclei of the individual cells are round with fine chromatin and nucleoli may or may not be present. Rosette is usually seen. Immunohistochemical analysis of Ewing Sarcomas reveals expression of vimentin and CD99 (MIC-2), with characteristic perinuclear staining. MIC-2 is a specific marker for Ewing Sarcoma/PNET [8]. Differential diagnosis of tumours involving the skull with adjacent bony erosion in children include metastatic neuroblastoma, Ewing Sarcoma/PNET, chordoma, lymphoma, rhabdomyosarcoma, osteosarcoma, meningioma, langerhan’s cell histiocytosis, desmoplastic small round cell tumour, plasmacytoma, and solitary metastasis. The differentiation between these can be done by histopathological examination including light microscopy and special stains. Immunohistochemistry is usually required for final diagnosis.
Data from retrospective analysis suggest that, Ewing Sarcoma can be better cured by surgery (with or without post-operative radiotherapy) than by radiotherapy alone. In cases where surgical excision is not possible, radiotherapy combined with chemotherapy can be given. Among chemotherapeutic agents, the most active substances are Doxorubicin, Cyclophosphamide, Ifosfamide, Vincristine, Dactinomycin, and Etoposide [9].
Conclusion
Primary Ewing Sarcoma of cranium should be considered as a differential in tumours extending intra-cranially with bony destruction. Prognosis of primary Ewing Sarcoma of skull with intra-cranial extension is poor and so early diagnosis by radiology, histopathology and immunohistochemistry is mandatory. In neurosurgical practice, primary Ewing’s Sarcoma of the cranial bones requires early aggressive management to achieve adequate long term remission and cure.
[1]. Desai KI, Nadkarni TD, Goel A, Muzumdar DP, Naresh KN, Nair CN, Primary Ewing’s sarcoma of the craniumNeurosurgery 2000 46:62-8. [Google Scholar]
[2]. Wang D, Guo Z, Multiple primary Ewing’s sarcomas in cerebral cranium of a child: a case report and review of the literatureInternational Journal of Clinical and Experimental Pathology 2015 8:7575-82. [Google Scholar]
[3]. Sim FH, Unni KK, Beabout JW, Dahlin DC, Osteosarcoma with small cells simulating Ewing’s tumourBone and Joint Surg 1979 61:207-15. [Google Scholar]
[4]. Singh P, Jain M, Singh DP, Kalra N, Khandelwal N, Suri S, MR findings of primary Ewing’s sarcoma of greater wing of sphenoidAustralas Radiology 2002 46:409-11. [Google Scholar]
[5]. Naama O, Ajja A, Gazzaz M, Albouzidi A, Belhachmi A, Asri A, Primary Ewing sarcoma of the skull base with cerebral extension. A case reportNeuroradiology 2007 4:689-98. [Google Scholar]
[6]. Garg A, Ahmad FU, Suri A, Mahapatra AK, Mehta VS, Atri S, Primary Ewing’s sarcoma of the occipital bone presenting as hydrocephalus and blindnessPediatric Neurosurg 2007 43:170-73. [Google Scholar]
[7]. Sim FH, Unni KK, Beabout JW, Dahlin DC, Osteosarcoma with small cells simulating Ewing’s tumourJournal of Bone and Joint Surgery 1979 61:207-15. [Google Scholar]
[8]. Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M, MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumours: evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumours from MIC2 expression and specific chromosome aberrationCancer 1991 67:1886-93. [Google Scholar]
[9]. David T, Mamot C, Krasniqi F, Metternich F, Prestin S, Multimodality Treatment in Ewing’s Sarcoma Family Tumours of the Maxilla and Maxillary Sinus: Review of the LiteratureSarcoma 2016 2016:3872768 [Google Scholar]