Malignant Granular Cell Tumour Presenting as a Paravertebral Mass in an Adolescent Male- A Rare Presentation of an Uncommon Tumour
Monika Singh1, Ajay Kr Singh2, Swasti Shubham3, Pratibha Maan4, Udit Chauhan5
1 Senior Resident, Department of Pathology, Dr. RML Hospital, Delhi, India.
2 Assistant Professor, Department of Pathology, KGMU, Lucknow, Uttar Pradesh, India.
3 Senior Resident, Department of Pathology, Dr RML Hospital, Delhi, India.
4 Senior Resident, Department of Pathology, Dr. RML Hospital, Delhi, India.
5 Senior Resident, Department of Radiology, GB Pant Hospital, Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Monika Singh, Room No -10 NRDH, GB Pant Hospital, Jawahar Lal Nehru Marg, New Delhi-110002, India.
E-mail: mona.nov10@gmail.com
Granular Cell Tumour (GCT), also known as Abrikossoff’s tumour is a rare neural tumour, mostly benign and solitary but rare malignant and multifocal occurrence are also reported. Location of tumour varies widely within body with tongue, skin and subcutaneous tissue being the most common sites. We report a case of malignant GCT in a 17-year-old male presented with a paravertebral swelling. Radiological and histopathological findings along with immunohistochemistry were of malignant GCT. We emphasize this case for its uncommon age and site of presentation in addition to invasive nature.
Abrikossoff’s tumour, Immunohistochemistry, Neural tumour
Case Report
A 17-year-old male presented with a slowly growing soft tissue mass of the left paravertebral region. Mass was asymptomatic ever since, its palpation. There was no associated pain or any changes of the overlying skin. There was no significant medical or family history of the patient. Routine blood investigations including haemogram were unremarkable. On clinical examination, the mass was palpable as a diffuse intra-abdominal swelling in the left lumbar region. A CECT abdomen was done which revealed a large abdomino-pelvic mass (approximately 10x6cm) with mild post-contrast enhancement and central hypodense non-enhancing portion consistent with necrosis. Lytic destruction of sacrum and iliac bone was seen on the left side with associated widening of neural foramen between first and second sacral vertebral bodies [Table/Fig-1]. A decision for excisional biopsy was made. Histological examination revealed a tumour composed of moderately pleomorphic large polyhedral cells with eccentrically placed nucleus having large prominent nucleoli. Tumour cells were having abundant granular eosinophilic cytoplasm. Mitotic count was < 2/10 High Power Field (HPF) [Table/Fig-2,3]. Immunohistochemistry was positive for S100 protein, CD68 and negative for Epithelial Membrane Antigen (EMA), desmin and myogenin [Table/Fig-4,5]. Histological features were suggestive of malignant GCT. A wide enbloc excision of the mass was planned with a work up of metastasis, but patient refused for the same and opted for alternative medicine and eventually lost for follow-up.
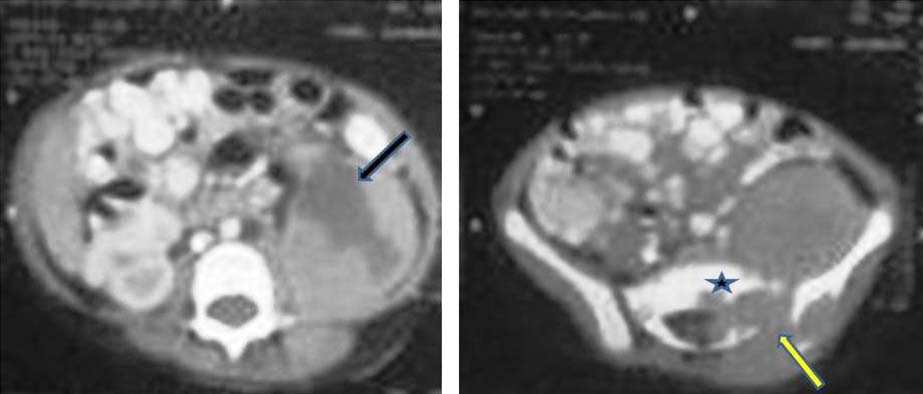
Axial Contrast Enhanced Computed Tomography (CECT) images revealing a large abdominopelvic mass extending from left renal fossa down till pelvis. The mass shows central hypodense areas consistent with necrosis (arrow in ‘a’). The mass is seen causing widening of neural foramen (arrow in ‘b’) in addition to causing lytic destruction of sacrum (* in ‘b’) and iliac bone (near arrow in ‘b’). The mass is seen to infiltrate into the paraspinal muscles. Necrosis, infiltration of paraspinal muscles and lytic destruction of bone suggested aggressive nature of mass.
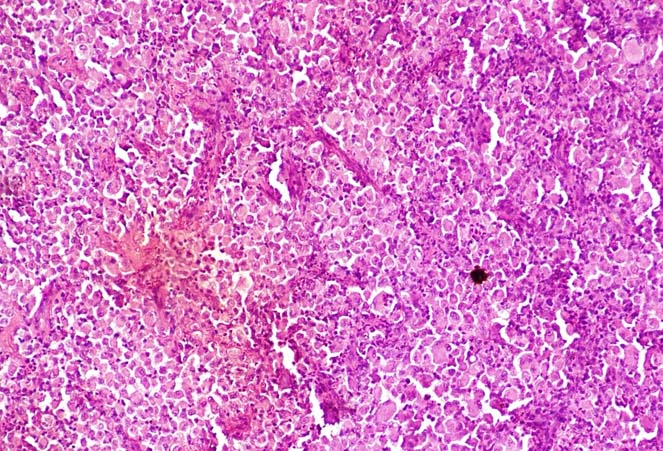
Histopathological examination revealed sheets of pleomorphic polyhedral cells with fine fibrous septa (H&E staining, X100).
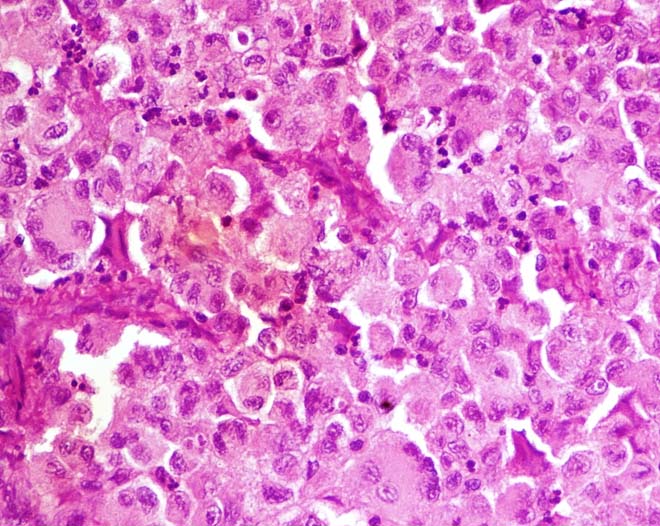
Tumour cells have round to oval eccentrically placed nuclei with vesicular chromatin and prominent nucleoli. Abundant granular cytoplasm due to lysosomes. Few large multinucleated cells are also seen (H&E staining, X400).
Tumour cells show strong cytoplasmic immunoreactivity for S-100 protein (Immunohistochemistry, X400).
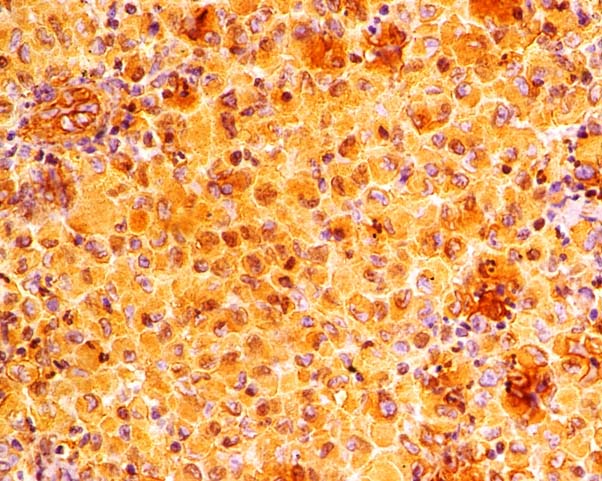
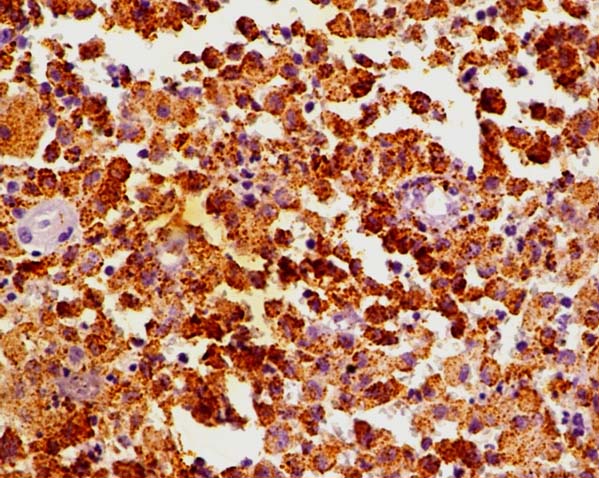
Tumour cells show strong cytoplasmic immunoreactivity for antigen CD68 (Immunohistochemistry, X400).
Discussion
Granular Cell Tumour (GCT), which is a rare soft tissue neoplasm of neural origin was first described by Abrikossoff in 1926 [1]. These neoplasms can arise in any part of the body but are particularly common in the tongue, subcutaneous tissue and skin [2]. It is most commonly seen in the third to the fifth decade of life and is rare in children. GCT is about twice as common in females as in males [3]. GCT have a controversial cell of origin, initially it was thought that, they arise from skeletal muscle [1]. Recently based on study of Kurtin and Bonin, it is believed that, these tumours arise from Schwann cells as schwanommas and GCT both show positive reactivity against monoclonal antibody KP-1, which is a marker against lysosomal glycoproteins [4]. GCT also shows cytoplasmic positivity against S-100 [5]. The vast majority of GCT pursue a benign course and constitute 0.5% of all soft tissue neoplasms with malignant forms representing less than 2% of GCT [6]. The benign GCT consists of polyhedral tumour cells with abundant granular eosinophilic cytoplasm with relatively small nuclei and prominent nuclei.
Fanburg Smith et al., have forwarded the following six morphological criterias for malignancy in GCT:
1) spindling of tumour cells;
2) increased nuclear to cytoplasmic ratio;
3) pleomorphism; 4) necrosis;
5) vesicular nuclei with large nucleoli; and
6) increased mitotic activity (>2 mitoses per 10 high-powered fields).
Neoplasms that meet three or more of these criteria are classified as malignant, whereas the atypical ones meet one or two criteria while benign ones display only focal pleomorphism [7].
The differential diagnosis of GCT include, Rhabdomyosarcoma, Alveolar soft part sarcoma, Hibernoma, Paraganglioma, Oncocytic neoplasms, Leiomyosarcoma, Epithelioid sarcoma and Metastatic renal cell carcinoma [8–10]. Rhabdomyosarcomas are also composed of polygonal shaped rhabdomyoblasts which show positivity for muscle markers in contrast GCT is positive for the S-100 protein and negative to desmin and myogenin [10]. Alveolar soft part sarcoma displays an alveolar pattern of arrangement of tumour cells showing marked nuclear pleomorphism in contrast to the sheets of polygonal cells in GCT. Hibernomas have vacuolated cytoplasm unlike granular cell tumour [10]. Paraganglioma can be differentiated by its location and positivity for chromogranin and synaptophysin [10]. Epithelioid sarcoma displays a background of necrosis and inflammatory cells with positivity for epithelial markers such as cytokeratins and Epithelial Membrane Antigen (EMA) [10]. Current treatment for malignant variant is wide local surgical excision with lymph node dissection. Role of radiation or chemotherapy is not definite [11].
Conclusion
Our case report highlights the uncommon presentation in an adolescent male of the rare malignant GCT along with the importance of Immunohistochemistry (IHC) panel used to rule out other differential diagnosis. Knowledge of which is of paramount importance for every practising pathologist in order to achieve a correct diagnosis and guide for adequate management of this condition.
[1]. Abrikossoff AI, Uber Myome, ausgehend von der quergestreifter willknerlicher muskulaturVirchows Arch Pathol Anat 1926 260:215-33. [Google Scholar]
[2]. Chaudry AP, Jacobs MS, SunderRaj M, Yamane GM, Jain R, Scharlock SE, A clinico- pathological study of 50 adult oral granular cell tumoursJ Oral Med 1984 39:97-103. [Google Scholar]
[3]. Liu TT, Han Y, Zheng S, Li B, Liu YQ, Chen YX, Primary cutaneous malignant granular cell tumour: a case report in China and review of the literatureDiagnostic Pathology 2015 10:113doi:10.1186/s13000-015-0357-2 [Google Scholar]
[4]. Kurtin PJ, Bonin DM, Immunohistochemical demonstration of the lysosome –associated glycoprotein CD68 (KP-1) in granular cell tumours and schwanomasHum pathol 1994 25:1172-78. [Google Scholar]
[5]. Mukai M, Immunohistochemical localization of S-100 protein and peripheral nerve myelin proteins (P2 protein, PO protein) in granular cell tumoursAnn J Pathol 1983 112:139-46. [Google Scholar]
[6]. Enzinger RM, Weiss SW, Granular cell tumour, in Enzinger RM, Wiess SW (eds)Soft tissue tumours 1995 ed 3St Louis, MOMosby:864-875. [Google Scholar]
[7]. Fanburg Smith JC, Meis Kindblom JM, Fante R, Kindblom LG, Malignant granular cell tumour of soft tissue. Diagnostic criteria and clinicopathologic correlationAm J Surg Pathol 1998 22:779-94. [Google Scholar]
[8]. Korambayil PM, Granular cell tumour of the handIndian J Plast Surg 2012 45:160-62. [Google Scholar]
[9]. Loncar B, Marjanovi K, Pauzar B, Staklenac B, Granular cell tumour clinically presented as lymphadenopathyColl Antropol 2010 34:261-63. [Google Scholar]
[10]. Sung JW, Choi JH, Imprint cytology of granular cell tumour: A case reportKorean Journal of Cytopathology 2007 8:170-74. [Google Scholar]
[11]. Batsakis JG, Manning JT, Soft tissue tumours: Unusual forms, OtolaryngolClin North Am 1986 19:659-83. [Google Scholar]