Case Report
A 46-year-old female with past medical history of bronchial asthma, on inhaled corticosteroids for the last 12 years and primary hypothyroidism, on thyroxin supplementation for five years. She was admitted for weakness in both ankles with bilateral foot drop (right>left) and hand paresthesias with grip weakness of two months duration, along with significant weight loss. Weakness was insidious in onset and gradually progressive over a period of time to the extent that at presentation, the patient was not able to walk and was brought on a wheel chair. On examination, she had bilateral foot drop, bilateral complete loss of touch, pain and temperature over superficial peroneal nerve distribution, bilateral upper limb dysasthesia in median and ulnar distribution, absent ankle jerk and high steppage gait. On investigation, the patient had a raised ESR (69 mm fall in first hour), positive Rheumatoid factor (RA) and C-Reactive Protein (CRP). Test for perinuclear anti-neutrophil cytoplasmic antibody (p-ANCA) was positive [Table/Fig-1]. Her routine investigation which included complete blood count, liver and renal function test, blood coagulation studies, electrolytes and urine examination were within normal limits. Serologic tests for Hepatitis B, Hepatitis C and HIV were negative. Anti-Nuclear Antibody (ANA) was negative. Chest radiograph revealed no abnormality. Nerve Conduction Study (NCS) revealed bilateral low amplitude Compound Muscle Action Potential (CMAP) in common peroneal nerve distribution and absent sural nerve Sensory Nerve Action Potential (SNAPs) bilaterally. A left sural nerve biopsy was done and it showed epineural vessels vasculitis with dense perivascular and transmural inflammatory infiltrate comprising of lymphocytes, plasma cells and histiocytes with evidence of fibrinoid necrosis and axonal breakdown [Table/Fig-2]; no granulomas were observed and uniform moderate loss of small and large myelinated fibres were seen-suggestive of vasculitis. She had no history of switching inhaled steroid medications to oral steroids or the use of leukotriene antagonists. Our patient was treated with Inj Methylprednisolone (1 gm IV infusion/day for five days) and cyclophosphamide pulse therapy (1 gm/m2 IV every month). She had a slow but sustained improvement over six months, with 90% improvement in upper limb symptoms and ankle dorsiflexors. She had complete remission of bronchial asthma, not requiring any more inhaled steroids after three months of immunosuppressive therapy. She was able to achieve a good quality of life and became independent for all activities of daily living at six months of therapy (from a previous wheel chair bound state). She has been switched to two monthly cyclophosphamide pulses after first six months based on good therapeutic response, which we plan to continue for one year. Thereafter, she will be followed up based on disease activity.
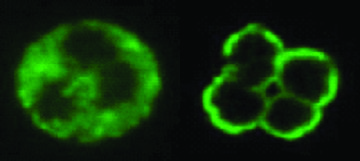
Immunofluorescence images of serum samples demonstrating various ANCA patterns in human neutrophils - c-ANCA shows a diffusely granular, cytoplasmic staining pattern and p-ANCA shows a perinuclear staining pattern.
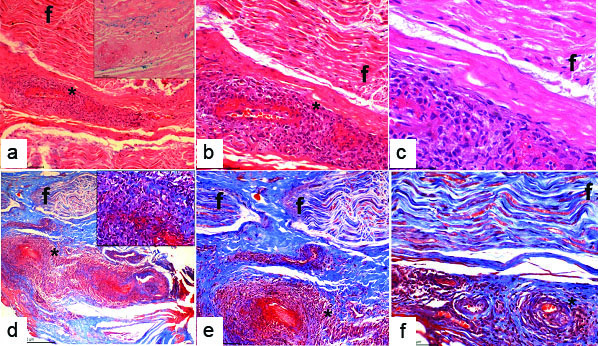
Sural nerve biopsy specimen: a) Nerve funicle (f) with epineureal vessel showing transmural and perivascular mononuclear cell infiltrates [H&E X 100], inset showing haemosiderin deposits which are Perl’s stain positive (Perl’s stain, X 200); b) Nerve funicle (f) with transmural and perivascular mononuclear cell infiltrates (H&E X 200); c) Nerve funicle (f) with fibrinoid necrosis (H&E X 400); d) Nerve funicle (f) with an epineureal vessel with mononuclear cell infiltrates. (Masson’s trichrome stain H&E X 100), Inset showing inflammatory cells and a few neutrophils (Masson’s trichrome X 400); e) Nerve funicle; (f) with vasculitis and necrosis (Masson’s trichrome stain H&E X 200); f) Microphotograph showing a nerve funicle (f) with an epineureal vessel displaying a transmural and perivascular mononuclear cell infiltrates with necrosis (Masson’s trichrome stain H&E X 400), evidence of axonal loss is seen.
Discussion
The ANCA-Associated Vasculitis (AAV) is a group of rare diseases, not often encountered in clinical practice and show characteristic inflammatory cell infiltration and blood vessel wall necrosis in histology. AAV is generally used to include primary vasculitis syndromes in which circulating ANCA against Proteinase 3 (PR3) called c-ANCA and Myeloperoxidase (MPO) called p-ANCA [Table/Fig-3] are commonly found. AAV syndromes include GPA, MPA and EGPA [1].
Chapel Hill consensus definitions (2012) for ANCA-associated vasculitis [5].
| ANCA- associated vasculitis (AAV) | Necrotizing vasculitis having few immune deposits or sometimes no immune deposits, characteristically involving small vessels. Usually associated with p-ANCA or c-ANCA and may be ANCA negative (not all patients show ANCA positivity). |
| GPA(Wegener’s granulomatosis) | Necrotizing granulomatous inflammation often showing respiratory tract involvement (upper and lower respiratory tract) and necrotising vasculitis of small to medium vessels Renal involvement characteristically shows necrotising glomerulonephritis. |
| EGPA(Churg-Strauss syndrome) | Necrotizing granulomatous inflammation which is eosinophil-rich frequently involving the respiratory tract, and necrotizing vasculitis of small to medium vessels. Clinically associated with asthma and eosinophilia. Presence of glomerulonephritis is associated with increased incidence of ANCA positivity. |
| MPA | Necrotizing vasculitis having few immune deposits or sometimes no immune deposits, characteristically involving small vessels. Necrotizing arteritis of small and medium arteries may be present. More commonly associated with necrotizing glomerulonephritis. No histological evidence of granulomatous inflammation. |
There is no definitive classification of systemic vasculitis and it remains controversial because of its varied aetiology which is largely unknown. Further, there is considerable overlap in the clinical presentation of the different vasculitic syndromes. The general way of classification is based on the size of dominant vessel and ANCA status. However, AAV is grouped separately since they involve small and medium-sized arteries which are frequently ANCA associated. Also, AAV is associated with increased risk of glomerulonephritis and respond adequately to immunosupression (cyclophosphamide) [2]. The probable reason is that the aetiology may not be related to immune complex formation unlike pure small-vessel vasculitis like Henoch-Schönlein purpura and cryoglobulinaemic vasculitis.
In 1990, the American College of Rheumatology (ACR) developed classification criteria for systemic vasculitis. They did not consider microscopic polyangiitis but considered Wegener’s granulomatosis, Churg–Strauss syndrome and polyarteritis nodosa. This classification criterion carried high sensitivity (82-87%) and specificity (87-92%) but the drawback is that it was not validated in individuals without vasculitis [3]. The Chapel Hill Consensus Conference (CHCC) in 1994 defined AAV which comprised microscopic polyangiitis but lacked the criteria to diagnose [4]. These were revised and updated in 2012 [Table/Fig-1] and a revised nomenclature introduced – granulomatosis with polyangiitis (Wegener’s granulomatosis) (GPA), MPA, and EGPA (Churg–Strauss syndrome) [5].
Granulomatosis with Polyangiitis (GPA)
GPA, previously known as Wegener granulomatosis, is an uncommon autoimmune disease, especially in Indian population. The disease spectrum includes necrotising granulomata of the respiratory tract (upper and lower), necrotising vasculitis involving various organs like eyes, throat, skin and frequently involves the kidneys causing focal glomerulonephritis. The most common clinical presentation is the upper airway disease. It is seen in upto 90% of patients and almost 70% of patients initially present with upper airway disease [6]. Destruction of the nasal septum results in the typical saddle-nose deformity. About 85% of patients develop sinusitis during the course of the disease. Involvement of larynx and trachea is usually asymptomatic, but may present with hoarseness or stridor [6].
Renal disease occurs in 18% of patients as part of initial presentation and subsequently seen in 77% of the affected individuals [7]. Renal involvement is not a common initial presentation but any renal involvement is associated with a significantly poor outcome. The clinical presentation includes renal failure as a sequele to rapidly progressive glomerulonephritis and is often life-threatening. Such conditions warrant urgent haemodialysis and immunosupression [7].
Pulmonary involvement is one of the main features of GPA, occurring in 45% at presentation and 87% during the course of the disease, with cough, haemoptysis and pleurisy being the common clinical symptoms [6]. The most frequent radiological features are pulmonary infiltrates, pulmonary haemorrhage and hilar lymphadenopathy [8].
Cutaneous manifestations occur in around 50% of patients, and include ulcers, palpable purpura, papules and nodules [8].
Neurological involvement is uncommon at presentation and presents as peripheral neuropathy or mononeuritis multiplex. Mostly the symptoms are subclinical and can be detected in ≤50% of patients in the course of illness [9].
Eye involvement occurs in almost half of the patients and any part of the eye can be potentially affected. The clinical manifestation may range from keratitis and scleritis to retinal vessel occlusion and optic neuritis [9]. Visual loss is reported in ≤8% of patients.
Musculoskeletal symptoms occur commonly and majority of patients have arthralgias and/or myalgias. Involvement of the joint causing true synovitis is not common and is seen in around 25% of patients [9].
Microscopic Polyangiitis (MPA)
The initial description of MPA was given by Kussmaul and Maier when they described a patient showing peri-arteritis with inflammatory necrosis of medium-sized arteries resulting in organ infarction. The term microscopic polyarteritis nodosa was first described by Wainwright and Davson in a group of patients who in addition to features of Polyarteritis Nodosa (PAN) also had segmental glomerulonephritis manifesting as rapidly progressive renal failure [10]. Presently, this syndrome is popularly known as Microscopic Polyangiitis (MPA). Patients with MPA have clinical presentation mimicking GPA patients and in initial stage of the disease distinction may not be easy.
Renal involvement is characteristic for MPA. The classical histological feature is a pauci-immune focal segmental necrotising glomerulonephritis which is also the typical feature seen in GPA. Respiratory involvement causing pulmonary haemorrhage is a common clinical finding (≤29%) [10].
The occurrence of Mononeuritis multiplex in MPA is uncommon (≤20%) similar to GPA. Also, eye and nasopharyngeal involvement are uncommon in MPA than in GPA (<30%) [6].
Eosinophilic Granulomatosis with Polyangiitis (EGPA)
The characteristic clinical features of EGPA include asthma which typically occurs late in the course of the disease associated with peripheral blood and tissue eosinophilia in addition to fever and systemic vasculitis [11]. The occurrence of asthma which precedes the onset of systemic features in almost all cases of EGPA distinguishes it from other forms of vasculitis especially GPA and MPA [12]. Histologically, EGPA exhibits necrotizing granulamatous vasculitis [5]. Renal disease is not common in EGPA unlike MPA and GPA where the characteristic clinical manifestation is renal disease. Pulmonary involvement (Chest radiograph showing reticulo-nodular infiltrates) is seen in ≤77% [12]. Cardiac disease is common (≤ 47%) and is an important cause of mortality, due to congestive heart failure, pericardial effusion and restrictive cardiomyopathy [11]. Unlike MPA and GPA, mononeuritis multiplex is a common (70%) feature of EGPA [11].
Diagnostic criteria for Eosinophilic Granulomatosis with Polyangiitis (EGPA) [11]: In 1990, the diagnostic criteria for EGPA were formulated by the ARA which included six criteria. Presence of four or more criteria establishes the diagnosis and yields a sensitivity of 85% and an even better specificity of 99.7%. The criteria are: 1) Asthma (wheezing, expiratory rhonchi); 2) Eosinophilia of more than 10% in peripheral blood; 3) Mononeuritis multiplex or polyneuropathy; 4) Pulmonary infiltrates (may be migratory or transient); 5) Paranasal sinusitis; 6) Histological evidence of vasculitis with extravascular eosinophils [11].
Our patient had late onset bronchial asthma for which she was under regular medication. She had mononeuritis multiplex involving the upper and lower limbs which was gradually progressive. However, there was no evidence of peripheral eosinophilia, paranasal sinus abnormality, pulmonary involvement or abnormal chest radiograph finding. Nerve biopsy was suggestive of vasculitis but without eosinophilic infiltrate. The clinical picture, evaluation and investigation do not meet the minimum four out of six criteria mentioned by ARA to term as EGPA. Also, there is no renal involvement to suggest MPA. The absence of necrotizing granuloma of the respiratory tract and renal involvement in the clinical course of the disease, rules out GPA as a possibility.
Hence, our patient is most likely an overlap between EGPA and MPA. The treatment for both these disorders is broadly similar, and the patient showed excellent remission with steroids and pulsed cyclophosphamide therapy [2].
Conclusion
ANCA-associated vasculitis is an uncommon disease entity of an unknown aetiology. Though there have been significant strides in understanding the pathogenesis of the illness, the exact aetiology still remain nebulous. The advent of immunosuppressive therapy has dramatically changed the outcome of ANCA-associated vasculitis which otherwise carried a high mortality. Treatment options include immunosuppressant therapy either as monotherapy (cyclophosphamide) or combination therapy (glucocorticoid plus cyclophosphamide). For patients who are refractory to the standard regimen, rituximab and interferon alfa are newer options. Our patient showed excellent remission with GC plus cyclophosphamide pulse therapy having 90% improvement in upper limb symptoms and ankle dorsiflexors. She had complete remission of bronchial asthma, and is now independent for all activities of daily living. She will need long term immunosuppressive therapy and follow up for disease activity.
[1]. Xiao H, Hu P, Falk R, Jennette J, Overview of the pathogenesis of ANCA-associated vasculitisKidney Diseases 2015 1(4):205-15. [Google Scholar]
[2]. Harper L, Morgan M, Walsh M, Hoglund P, Westman K, Flossmann O, Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: Long-term follow-upAnnals of the Rheumatic Diseases 2011 71(6):955-60. [Google Scholar]
[3]. Fries J, Hunder G, Bloch D, Michel B, Arend W, Calabrese L, The American College of Rheumatology 1990 criteria for the classification of vasculitis: SummaryArthritis & Rheumatism 1990 33(8):1135-36. [Google Scholar]
[4]. Jennette J, Falk R, Andrassy K, Bacon P, Churg J, Gross W, Nomenclature of Systemic VasculitidesArthritis & Rheumatism 1994 37(2):187-92. [Google Scholar]
[5]. Jennette J, Falk R, Bacon P, Basu N, Cid M, Ferrario F, 2012 Revised International Chapel Hill Consensus Conference Nomenclature of VasculitidesArthritis & Rheumatism 2012 65(1):1-11. [Google Scholar]
[6]. Harabuchi Y, Kishibe K, Komabayashi Y, Clinical manifestations of granulomatosis with polyangiitis (Wegener’s granulomatosis) in the upper respiratory tract seen by otolaryngologists in JapanClin Exp Nephrol 2013 17(5):663-66. [Google Scholar]
[7]. Katada A, Granulomatosis with polyangiitis (Wegener’s granulomatosis)Larynx Japan 2012 24(2):74-79. [Google Scholar]
[8]. Watts RA, Dharmapalaiah C, The role of biologics in ANCA-associated vasculitisModern Rheumatology 2011 22(3):319-26. [Google Scholar]
[9]. Langford CA, Fauci AS, The Vasculitis Syndromes. In: Kasper DL, Fauci AS, Hauser, Longo, Jameson, Loscalzo, editorsHarrison’s principles of internal medicine 2015 19th edNew York (NY)The McGraw-Hill Companies, Inc:2179-2193. [Google Scholar]
[10]. Villiger P, Guillevin L, Microscopic polyangiitis: Clinical presentationAutoimmunity Reviews 2010 9(12):812-19. [Google Scholar]
[11]. Gioffredi A, Maritati F, Oliva E, Buzio C, Eosinophilic granulomatosis with polyangiitis: An overviewFront Immunol 2014 5:549 [Google Scholar]
[12]. Uematsu H, Takata S, Sueishi K, Inoue, H, Polyangiitis overlap syndrome of granulomatosis with polyangiitis (Wegener’s granulomatosis) and eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome)BMJ Case Reports 2014 2014:bcr2013010195 [Google Scholar]