Neuroblastoma is the most common solid malignant neoplasm in the paediatric age group; accounting for 7% of all childhood malignancies, but neuroblastoma in adult, is a rare occurrence, finding mention in aeons of medical literature with an overall incidence of 1 in 10 million adults/year. We report the case of a 24-year-old male patient presenting with the complaints of progressive abdominal distention of 2 months duration. Multiple Detector Computed Tomography (MD-CT) of abdomen revealed a huge enhancing mass in the retroperitoneum abutting the left kidney, and a subsequent CT-guided biopsy and immunohistochemistry confirmed the diagnosis of ganglioneuroblastoma. The patient was administered 3-cycles of neo-adjuvant chemotherapy with ifosphamide, carboplatin and etoposide (ICE-chemotherapy) and subsequently 3 more cycles of chemotherapy post surgery, followed by radiation. The rarity of this neoplasm in adults accounts for the lack of standardized staging and treatment protocols and the dismal prognosis even with aggressive multimodal treatment. We report this case because of its extreme rarity, and its tendency for capricious behavior.
Case Report
A 24-year-old male, presented to the oncology outpatient department of Saifee hospital with the complaints of abdominal distention, pain and significant weight loss since 2 months. Clinical examination revealed a mass in the left inferior quadrant of abdomen and lumbar region.
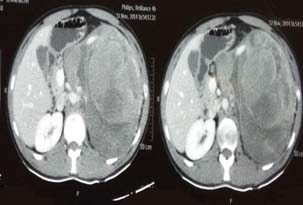
Multiple Detector Computed Tomography (MD-CT) of abdomen [Table/Fig-1] revealed a large heterogeneously enhancing mass in the left half of abdomen measuring 16 x 12 x 18.8 cm in dimensions, and extending from left hypochondrium to the lower lumbar region displacing the kidney. A 99mTc-HYNIC TOC scan [Table/Fig-2] revealed a heterogeneous mass with diffusely increased tracer uptake along with a small focus of increased uptake in the left supraclavicular region suggestive of solitary metastatic disease.
CT-Abdomen revealing a large enhancing mass in the left retroperitoneum.
99mTc-HYNIC TOC scan reveals a heterogeneous mass with diffusely increased tracer uptake and a foci of increased uptake in the left supraclavicular region.

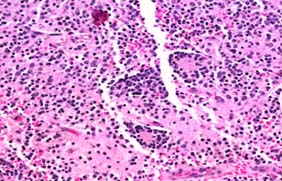
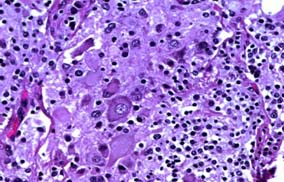
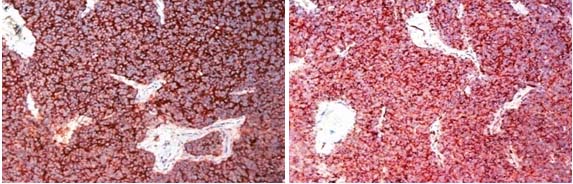
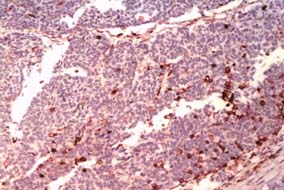
CT-Guided biopsy of the mass revealed small round blue cells forming rosettes with scant cytoplasm and scattered pale pink staining neurofibrillary stroma along with scattered ganglion cells [Table/Fig-3,4]. Immunohistochemically the tumour cells were positive for chromagranin A, synaptophysin [Table/Fig-5,6] but negative for Leucocyte Common Antigen (LCA) [Table/Fig-7], thus confirming the diagnosis of neuroblastoma. A staging bone marrow aspiration and biopsy revealed an uninvolved marrow. A 24 Hour urine VMA and metanephrine levels were unremarkable. The patient received 3-cycles of neo-adjuvant ICE-chemotherapy regimen, subsequently operated and administered 3 more cycles of ICE chemotherapy post surgery, followed by radiation. Follow-up at 3 months from the completion of radiotherapy, showed the patient was in fine health. He was being planned for follow-up Metaiodobenzylguanidine scan (MIBG scan)/Positron Emission Tomography (PET scan) scan at 6-months.
Section revealing small round cells with scant cytoplasm forming rosettes scattered over pink schwannian stroma. (H&E 20x).
Section revealing large ganglion cells & over a fibrillary and necrotic background (H&E 40x).
Sections reveal cytoplasmic positivity for Chromogranin A & Synaptophysin (20x).
Tumour was negative for LCA (20x).
Discussion
Neuroblastoma arises from the primitive neural crest cells of the adrenal medulla and neural cell rests of the sympathetic ganglia [1]. It was first described by Dr. Rudolf Virchow as “Gliomas of the abdominal cavity” in 1864 [2]. It was Dr. Homer Wright who conjectured the neural origin of this tumour based on histological rosettes it formed in the bone marrow [3].
Incidence and Demographics
Neuroblastomas are the most common solid extra cranial tumour of childhood, accounting for > 7% of all childhood malignancies (<15 years) [4,5] and has cumulative incidence of 2.26 cases per million inhabitants per year [6,7] with 60% of the cases being diagnosed within first 2 years of life [8,9]. But its incidence in adults is exceedingly rare with a predicted incidence of 1 case per 10 million adults per year with < 100 cases reported in medical literature [10,11].
A 20 year (Jan 1982 to Jan 2001) retrospective database review done at the Instituto Nazionale Tumouri of Milan by Podda MG et al., yielded a total of 27 patients over the age of 12 years [12] and the median age of presentation was 17 years (range 12-69 years).
In a similar 18 year (Jan 1994-Sept 2012) database review by Conter HJ et al., at the MD Anderson Cancer Center yielded 230 patients with the diagnosis of neuroblastoma/ganglioneuroblastoma, of which 118 (51.3%) were adults, and the median age at presentation was 47 years (range 18-82 years) [13].
Clinical Features
Podda MG et al., reported the most common site for primary as retroperitoneum (70.3%), followed by adrenal (37.03 %), abdominal (33.3%), thorax (18.5 %), and single case each of pelvis and thorax [12]. Conter HJ et al., reported the primary sites as head and neck in 82 (69.5%), followed by genito urinary tract in 13 (11.01%), soft tissue in 9 (7.62%), chest cavity in 5 (4.23%), and CNS in 4 (3.38%) cases respectively [13].
The most common presenting complaints are abdominal mass, abdominal pain, weakness, and symptoms due to distant metastases like back ache, bone marrow failure, spinal compression, and hypertension [8,11–13].
Adults commonly present with an unfavourable histology and at advanced stages of disease, with nearly one third having distant metastases on presentation, but bizarrely a lower frequency of bone marrow involvement as corroborated in the study by Podda MG et al., (59.25%), & Conter HJ et al., (37.2%) [12–14].
Treatment & Prognosis
Reviewed of & found Podda MG et al., 27 patients aged 12-69 years [12], the treatment protocol followed was Stage I-surgery only, Stage II-surgery followed by radiotherapy, Stage III-Chemotherapy consisting of 6 cycles of cisplatin & etoposide alternating with adriamycin, cyclophosphamide and vincristine and local therapy after 6th course consisting either of radiotherapy or surgery. Consolidation with radiation followed by 4 additional courses of chemotherapy. Stage IV-treatment is essentially same as stage III, except for the addition of ifosphamide to the cycles, followed by consolidation phase with 10 Gy HBI, or autologous stem cell rescue, local therapy with surgery or radiation was scheduled after the 4th cycle.
With above treatment protocol 5-year overall survival rate was 83% for stage I & II disease and 28% for patient with stage III & IV disease, but all patients with stage IV disease relapsed and died due to disease progression. The overall survival at 5 & 10 years was 40% and 20% respectively [12].
Podda MG et al., reported an absence of MYCN amplification in all the 5 cases evaluated for MYCN mutation which occurs in 20-40% of high risk paediatric tumours and correlates with an aggressive course [12]. Adult tumours on the other hand are conspicuous by the absence of mutated MYCN, suggesting a different biological process in their evolution, thus implying for different staging, treatment and prognosification criteria [11,12].
Conter HJ et al., reviewed of 118 cases at the MD Anderson Cancer Center and found that the treatment protocol for adults was [13]:-
Stage L1- The combination of surgery and radiation demonstrated the best progression free survival (median 11.1 months) compared to surgery, radiation alone or local therapy. The overall survival in this group was 18.1 years.
Stage L2- Surgery and radiation resulted in a median progression free survival of 5.2 months and addition of chemotherapy was not associated with an improved survival. The overall survival in this group was 9.8 years.
Stage M- The most common chemotherapy regimen employed was cisplatin and etoposide alternating with cyclophosphamide, vincristine and carboplatin. The median overall survival of patients with M-stage disease was 1.6 years.
Our patient was classified as stage 4 of International Neuroblastoma Staging System (INSS) [15] in view of distant nodal metastasis with Intermediate Grade Mitosis-Karyorrhexis Index (MKI) of 2-4%, & absent MYCN mutation [15].
International neuroblastoma risk group (INRGSS) staging with histological category, tumour grade, n-MYC mutations, 11q aberration and tumour ploidy has been shown to best risk stratify patients and help plan the next course of therapy [12–15].
Conclusion
Neuroblastoma in adults is a rare occurrence with insidious onset and dismal prognosis especially in advanced cases, with sparse data available on the natural history, genetic signature, treatment protocols and their outcomes, further research and co-operation between researchers and establishment of tissue banks, registries and enrollment of patients into novel treatment protocols will surely further our understanding of this disease.
[1]. Brodeur GM, Neuroblastoma: Biological insights into a clinical enigmaNature reviews Cancer 2003 3(3):203-16. [Google Scholar]
[2]. Virchow R, Hyperplasie der zirbel und der nebennieren [Hyperplasia of the pineal and suprarenal glands]Die krankhaften geschwulste 1864 2:1864-65. [Google Scholar]
[3]. Wright HJ, Neurocytoma or Neuroblastoma; A kind of tumour not generally recognizedJ Exp Med 1910 12:556-61. [Google Scholar]
[4]. Kaye JA, Warhol MJ, Kretschmer C, Landsberg L, Frei E, Neuroblastoma in adults-Three case reports and review of literatureCancer 1986 58:1149-57. [Google Scholar]
[5]. Woods WG, Gao RN, Shuster JJ, Robison LL, Bernstein M, Weitzman S, Screening of infants and mortality due to neuroblastomaNew Engl J Med 2002 346:1041-46. [Google Scholar]
[6]. Gurney JG, Ross JA, Wall DA, Bleyer WA, Severson RK, Robison LL, Infant cancer in the US: Histology specific incidence and trends,1973-1992J Paediatr Hematol Oncol 1997 19:428-32. [Google Scholar]
[7]. Davis S, Rogers MA, Pendergrass TW, The incidence and epidemiology characteristics of neuroblastomas in the United States AmericanJ Epidemiol 1987 126:1063-74. [Google Scholar]
[8]. Abdullah BB, Seehra N, Zoheb M, Nausheen N, Ahmed A, Rare occurrence of neuroblastoma in young adult-A diagnostic challengeInternal Med: Open access 2:106doi:10.4172/2165-8048.1000106 [Google Scholar]
[9]. Soares PBM, Filho SQ, De Souza WP, Neuroblastoma in an adult: Case reportRev Med Chile 2010 138:1131-34. [Google Scholar]
[10]. Siegel R, Naishadham D, Jemal A, Cancer statistics 2012CA: Cancer journal for clinicians 2012 62(1):10-29. [Google Scholar]
[11]. Rogowitz E, Babiker HM, Kanaan M, Millius RA, Ringenberg QS, Bishop M, Neuroblastoma of the elderly, an oncologist’s nightmare: Case presentation, literature review, and SEER database analysisExperimental Hematology & Oncology 2014 3:20 [Google Scholar]
[12]. Podda MG, Luksch R, Polastri D, Gandola L, Piva L, Collini P, Neuroblastoma in patients over 12 years old: A 20 year experience at the Instituto Nazionale Tumouri of MilanTumouri 2010 96(5):684-89. [Google Scholar]
[13]. Conter HJ, Gopalakrishnan V, Ravi V, Ater JL, Patel S, Araujo DM, Adult versus Paediatric neuroblastoma: The MD Anderson Cancer Center ExperienceSarcoma 2014 2014(375151):6pages. doi:10.1155/2014/375151 [Google Scholar]
[14]. Conte M, Parodi S, DeBernardi B, Milanaccio C, Mazzocco K, Angelini P, Neuroblastoma in adolescents: The Italian experienceCancer 2006 106:1409-17. [Google Scholar]
[15]. Brisse HJ, McCarville MB, Granata C, Krug KB, Wootton-Gorges SL, Kanegawa K, Guidelines for imaging and staging of neuroblastic tumours: consensus report from the International Neuroblastoma Risk Group ProjectRadiology 2011 261(1):243-57. [Google Scholar]