Case Report
A seven-year-old boy presented with pruritic erythematous rashes over his face since past 6 week with complain of not being able to sit, stand or take turns in bed and severe myalgia since past 4 week. Bilateral flexion deformity at knee, ankle, elbow, hip, wrist and interphalangeal joint with dysphagia for solid food was present since 2 weeks. There was no history of similar complaints in past. There was no complain of any trauma, fever, photosensitivity, change in voice or vision, breathlessness, abnormal behaviour and altered urinary or bowel habit. The child was born of non-consanguineous marriage and family history was not significant.
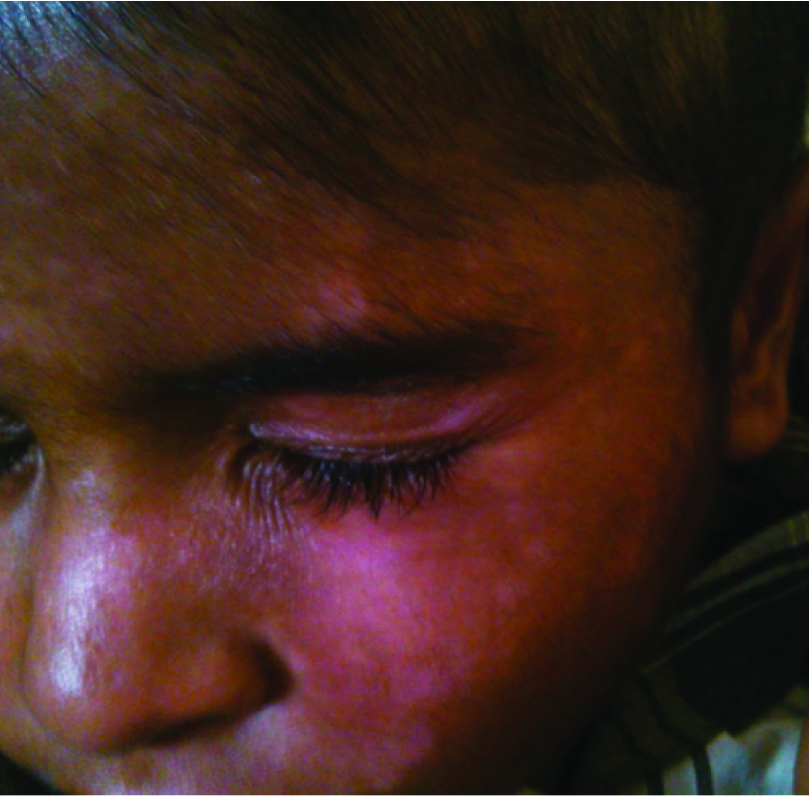
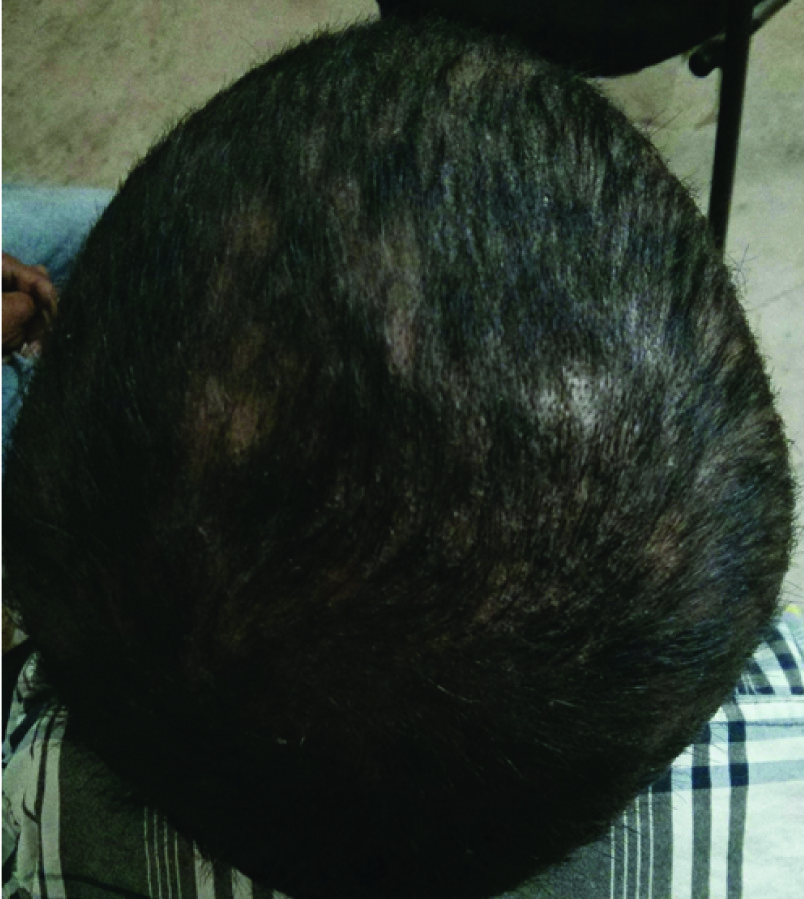
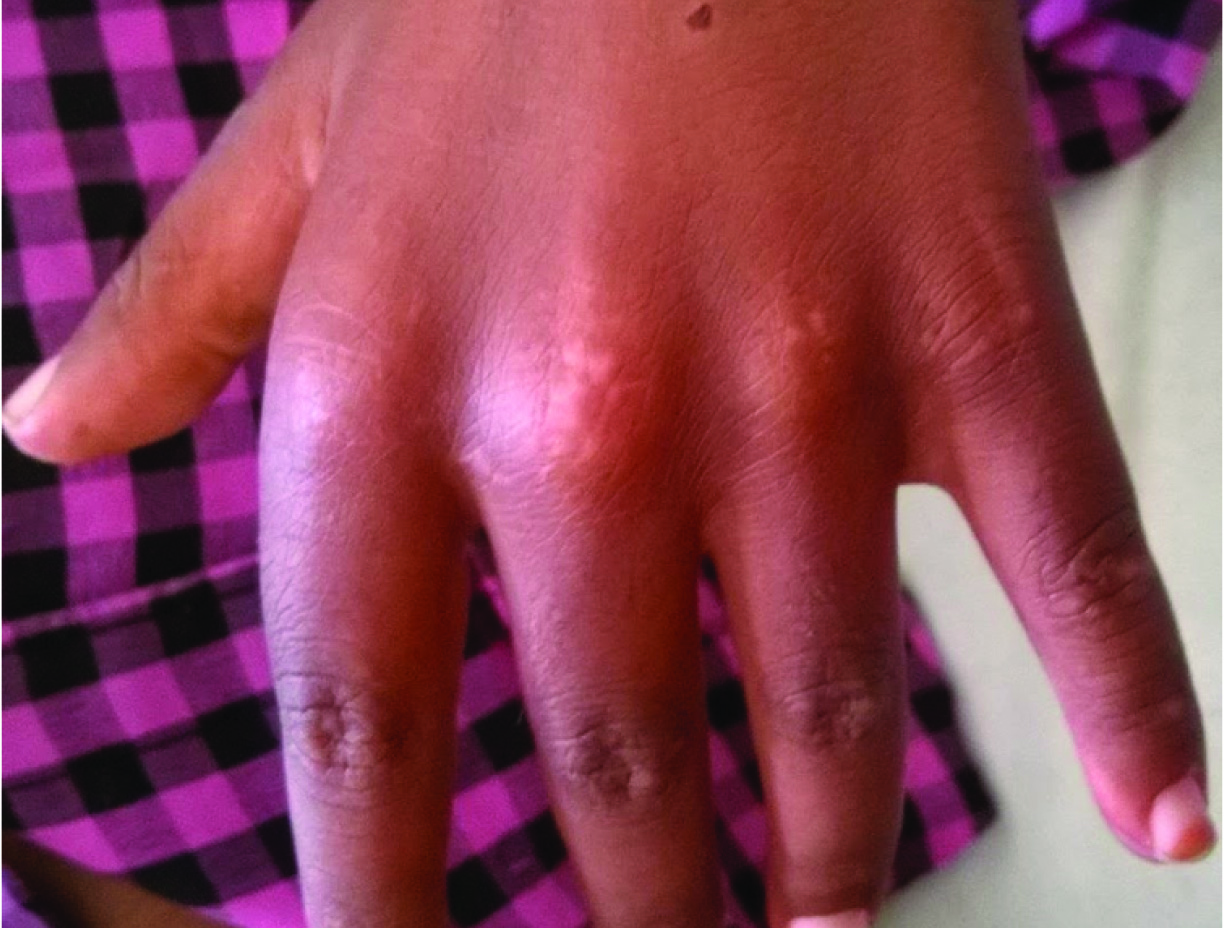
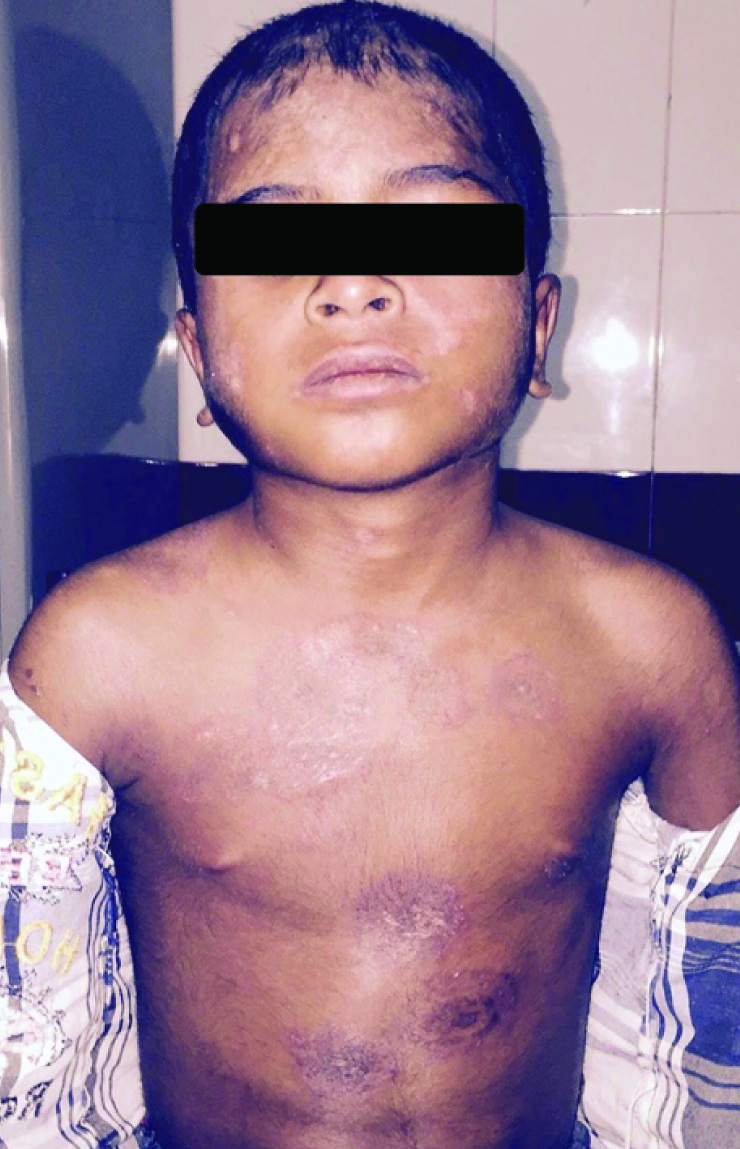
On physical examination his weight was 22kg, height was 122cm and a Body Mass Index (BMI) in 14.7Kg/m2. His vital signs showed body temperature 38.5°C, blood pressure 110/70mmHg, heart rate 74 beats per minute and respiratory rate 20 breaths per minute. He had ill-defined erythematous macular rashes over face along with swelling and reddish-violaceous rash over upper eyelids consistent with heliotrope rash [Table/Fig-1]. He had scaly scalp, thinned hair and hair loss [Table/Fig-2]. The skin over the metacarpal and proximal interphalangeal joints had thickened pale red papules suggestive of Gottron papules [Table/Fig-3]. Child also had hyper pigmented scaly plaques over trunk and extremities [Table/Fig-4]. Severe muscle tenderness over extremities and back was present. Bilateral Flexion contractures involving almost every joint of upper and lower extremities were present; therefore, deep tendon reflexes and power in the extremities could not be tested. Power of neck flexor and extensor was 2/5 and abdominal muscle weakness was present. Superficial reflexes, plantar response and sensory examination when performed were found to be normal. Other systemic examinations, which were done, were found to be inconspicuous.
Heliotrope rash on upper eyelid.
Scalp showing scales, thin hair and signs of hair loss.
Gottron papuels on skin above the metacarpals and proximal interphalengeal joints.
Hyperpigmented scaly plaques on skin above abdomen and extremities.
Investigations performed showed haemoglobin of 13.4gm/dl, total leukocyte count of 14400cells/mm3 and platelets 4.8lac cells/mm3. The peripheral smear showed thrombocytosis with neutrophilia and no evidence of abnormal cells. Erythrocyte Sedimentation Rate (ESR) was 40mm/hour, Lactate Dehydrogenase (LDH) was 970 U/L and C-Reactive Protein (CRP) level was 3.4mg/dl. Antinuclear Antibodies (ANA) and rheumatoid factor were negative. Creatine Phosphokinase (CPK) was 9865U/l, Alanine Transaminase (ALT)= 219U/L, Aspartate Transaminase (AST)=250U/L.
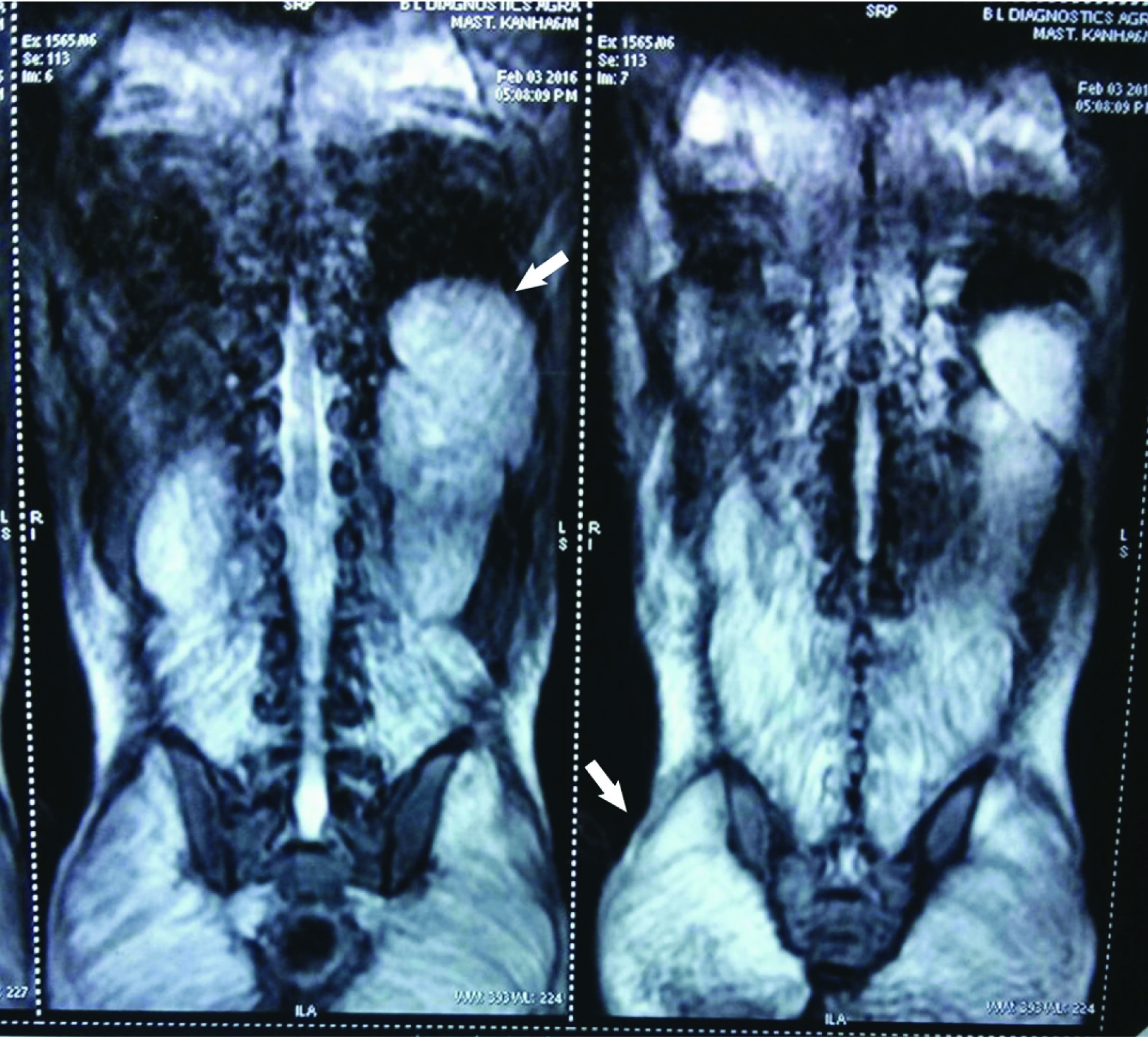
Magnetic Resonance Imaging (MRI) showed diffuse hyperintensity on FAT SAT T2 and T2 weighted images of back muscles, bilateral psoas, gluteal and pelvic muscle suggestive of myositis [Table/Fig-5]. Electrocardiogram (ECG) showed prolongation of PR interval with decreased heart rate (74beats/min). Chest X-ray and ECHO were normal.
Magnetic Resonance Imaging (MRI) Images: Showing diffuse hyper-intensity on FAT SAT T2 in the back muscles: Illio-psoas muscle (left arrow) and also involving the gluteus maximus muscle (right arrow).
Based on clinical features, laboratory investigation and MRI the diagnosis of Juvenile dermatomyositis was made. Patient was given methylprednisolone 30mg/kg/day intravenously for three days, oral steroids (2mg/kg/day), topical steroids, calcium and vitamin D supplements. Oral steroids were continued for two months at the dose of 2mg/kg/day, with the dose being now tapered off. After this treatment muscle weakness was improved day by day. On the 15th day of this treatment, proximal upper and lower, neck and shoulder motor extremity strength was improved dramatically but the contractures were still present in ankle, knee and elbow, and patient used support to walk. Physiotherapy is still continued for these contractures and improvement is being noticed. The peripheral blood smear revealed marked improvement in the neutrophil count. His ECG reports were normal too with no signs of prolong PR interval or bradycardia. Patients cardiac manifestation improved without any specific drug given for his this manifestations. Patient is still on follow-up and has shown no sign of any deterioration or recurrence.
Discussion
Juvenile Dermatomyositis (JDM) is a rare autoimmune disease but in paediatric population it is the most common inflammatory myopathy with unknown aetiology. Unlike adults, pure polymyositis without dermatological changes is uncommon in children. It can also involve numerous other organ systems, with significant mortality from cardiovascular, gastric and pulmonary sequelae of the disease [1].
In the United States, incidence of JDM ranges from 2.5-4.1 cases per million per year and females are affected more often than males, with a ratio of 2.3:1 [2]. The median age of onset of JDM is 6.8years in girls and 7.3years in boys [3,4]. In India, JDM accounts for about 2.5% of the paediatric rheumatology cases, mean age at diagnosis is 7.52 years and there is slight male preponderance [5-7].
Dermatomyositis has been recognized since 1887 and while it is still comparatively rarely observed it is of ever growing interest. There are comparatively few studies reported in paediatric age group of this disease. Unverricht was the first to recognize the association of this characteristic lesion of skin and muscle tissue and it was in 1891, he gave the disease its present name of dermatomyositis. Before that Wagner in 1863 had described this disease, which he apparently mentioned a case of “a rare muscular disease”. Later, Polain published a case report on dermatomyositis in 1875 and described it as “an atypical case of chronic glanders.”
JDM cases generally present as muscle weakness (90-100%), Skin lesions (85-100%), Erythematous rash of malar/facial area (42-100%), Heliotrope rash of eyelids (66- 83%), Gottron papules (57-91%), Muscle pain and tenderness (30-83%), Dysphagia or dysphonia (13-40%), Joint contractures (26-27%) and Cardiac involvement (0-3%) [8]. Scaly scalp or diffuse hair loss, in 40% of the cases [9]. Cardiac involvement is rare but heart murmurs, pericarditis and Electrocardiogram (ECG) abnormalities can be seen in JDM [10].
Though often not considered important but cardiac pathologies in JDM are one of the most common cause of mortality in these patients. The cardiac involvement could be either clinically overt or silent ranging in between 9 to 72% respectively [11]. Cardiac involvement as a cause of death in myositis was reported in 10–20% of cases [12]. This figure is rather not accurate as large epidemiologic studies of disease on JDM are rare. Common ECG abnormalities observed in dermatomyositis patients include: atrial and ventricular arrhythmias, bundle branch block, A-V blocks, high-grade heart block, prolongation of PR-intervals, ventricular premature beats, left atrial abnormality, abnormal Q-waves as well as non-specific ST-T wave changes.
The most common initial laboratory changes seen in JDM are in erythrocyte sedimentation rate, lactate dehydrogenase, and aspartate aminotransferase [13]. Diagnosis is made with the help of thorough history and physical examination supplemented by additional tests such as those of muscle enzymes, especially aldolase, LDH, ALT, AST and Creatine Kinase (CK). Apart from that test such as muscle biopsy, skin biopsy, electroneuromyography, muscle ultrasound and magnetic resonance imaging also aid in making the diagnosis [14]. MRI with T2- weighted fat suppression and Short Tau Inversion Recovery (STIR) is convenient in ruling out the differentials and making the diagnosis quickly as it reveals oedema in the tissue, which is a marker of muscle inflammation [15].
In the present case, we had symmetrical proximal muscle weakness and severe myalgia of lower limbs and initially which progressed to upper limb and trunk muscles associated with facial rash, heliotrope rash, gottron papules, contractures. On investigation elevated CRP, ESR, SGOT, SGPT and CPK was noted. MRI revealed myositis and ECG revealed PR interval prolongation suggestive of first degree AV block.
As per new criteria for diagnosis of JDM, this case is definitive JDM [16]. Changes in clinical practice over time have resulted in many clinicians using non-invasive techniques, which are beneficial to the point of view of the patient, such as MRI rather than using technique such as Electromyography (EMG) and muscle biopsy for making the diagnosis of JDM [17].
The co-relation of cardiac manifestations in dermatomyositis with overall severity of the disease is also contentious, but cardiovascular manifestations do constitute a major cause of death in these patients. This could either be due to underestimation of cardiac involvement in patient with dermatomyositis or the cardiac involvement is rather not addressed in the early phase of the disease. To address these questions controlled studies with carefully characterized patients are required.
Further research studies are needed to ascertain the prognostic significance of aggressive immunosuppressive therapy of patients with subclinical cardiac disease. Work is also desired in the area of better laboratory, imaging and serologic testing to identify patients who are at risk for the worst cardiovascular complications. At this point, based on many body of studies reviewed here and elsewhere, it is imperative that physicians treating JDM patients should undergo a routine cardiovascular risk assessment at the onset of diagnosis. Appropriate diagnostic and monitoring tests should also be inculcated into practise on those patients in which screening history or examination is suggestive of cardiac involvement.
The treatment of JDM consists of daily corticosteroids treatment along with non-steroidal immunosuppressant in adjunct. For patients with severe, refractory or corticosteroid-dependent disease, second line therapy consisting of combination of I.V gamma globulin, Cyclosporine and Azathioprine is administered. Biologic agents are the new drugs, which are emerging for the treatment of JDM, supported to date primarily by various clinical studies and are considered as the third line agents. It includes drugs such as Cyclophosphamide, Tacrolimus, Rituximab, Myclophenolate Mofetil and Anti-tumor Necrosis Factor alpha agents [18]. The prognosis of the disease depends mainly on the age of onset, its course, any underlying cause, and steroid responsiveness. Though according to some researchers young age in JDM is associated with poor prognosis, in our case it was not found to be true as the patient showed marked improvement with steroid therapy with no signs of recurrence. Further the subclinical cardiac manifestation was also found to be resolved on follow-up.
Conclusion
The patient of JDM have been shown to have a reduced vagal response to the heart which is associated with elevated inflammatory markers, active disease and decreased myocardial function. Further studies are needed to determine whether this subclinical cardiac association is also associated with morbidity or mortality in patients with JDM. Since past ten years, the prognosis of JDM has improved more and more due to the early diagnosis and the adequate medications consisting primarily of immune suppression. However, the presence of systemic manifestations and the short and long term side effects of the treatment may adversely affect morbidity and mortality. Therefore, we still need to develop a much effective way for long-term beneficial treatment of JDM and factors which would lead to better prognosis, need to be evaluated.
[1]. Kim S, El-Hallak M, Dedeoglu F, Zurakowski D, Fuhlbrigge RC, Sundel RP, Complete and sustained remission of juvenile dermatomyositis resulting from aggressive treatmentArthritis Rheum 2009 60:1825-30. [Google Scholar]
[2]. Mendez EP, Lipton R, Ramsey-Goldman R, Roettcher P, Bowyer S, Dyer A, US incidence of juvenile dermatomyositis, 1995-1998: Results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases RegistryArthritis Rheum 2003 49(3):300-05. [Google Scholar]
[3]. Symmons DP, Sills JA, Davis SM, The incidence of juvenile dermatomyositis: Results from a nation-wide studyBr J Rheumatol 1995 34(8):732-36. [Google Scholar]
[4]. Pachman LM, Lipton R, Ramsey-Goldman R, Shamiyeh E, Abbott K, Mendez EP, History of infection before the onset of juvenile dermatomyositis: Results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research RegistryArthritis Rheum 2005 53(2):166-72. [Google Scholar]
[5]. Chowdhary V, Wakhlu A, Agarwal A, Misra R, Outcome in juvenile dermatomyositisIndian Paediatr 2002 39:931-35. [Google Scholar]
[6]. Singh S, Bansal A, Twelve years experience of juvenile dermatomyositis in NorthRheumatol Int 2006 26(6):510-15. [Google Scholar]
[7]. Chickermane PR, Mankad D, Khubchandani RP, Disease patterns of juvenile dermatomyositis from Western IndiaIndian Paediatr 2013 50:961-63. [Google Scholar]
[8]. Keitel W, Wolff HP, Originators and eponyms of dermatomyositis : Ernst Leberecht Wagner (1821-1888) and Heinrich Unverricht (1853-1912)Z Rheumatol 2016 75(4):429-34. [Google Scholar]
[9]. Rider LG, Miller FW, Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathiesJAMA 2011 305(2):183-90. [Google Scholar]
[10]. Kasteler JS, Callen JP, Scalp involvement in dermatomyositis. Often overlooked or misdiagnosedJAMA 1994 272(24):1939-41. [Google Scholar]
[11]. Dourmishev LA, Dourmishev AL, Pharmacologic agents in the treatment of DM. In: Dourmishev LA, Dourmishev AL, editorsDermatomyositis advances in recognition, understanding and management 2009 1st edBerlinSpringer:297-322. [Google Scholar]
[12]. Zhang L, Wang GC, Ma L, Zu N, Cardiac involvement in adult polymyositis or dermatomyositis: A systematic reviewClin Cardiol 2012 35(11):686-91. [Google Scholar]
[13]. Danko K, Ponyi A, Constantin T, Borgulya G, Szegedi G, Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: A longitudinal study of 162 casesMedicine 2004 83:35-42. [Google Scholar]
[14]. Peloro TM, Miller OF, Hahn TF, Newman ED, Juvenile dermatomyositis: A retrospective review of a 30-year experienceJ Am Acad Dermatol 2001 45(1):28-34. [Google Scholar]
[15]. Mahesh A, Panchapakesa Rajendran C, Rukmangatharajan S, Rajeswari S, Vasanthy N, Parthiban M, Juvenile dermatomyositis-clinical and laboratory profileJ Indian Rheumatol Assoc 2005 13:4-7. [Google Scholar]
[16]. Chan WP, Liu GC, MR imaging of primary skeletal muscle diseases in childrenAJR Am J Roentgenol 2002 179(4):989-97. [Google Scholar]
[17]. McCann LJ, Kirkham JJ, Wedderburn LR, Pilkington C, Huber AM, Ravelli A, Development of an internationally agreed minimal dataset for Juvenile Dermatomyositis (JDM) for clinical and research useTrials 2015 16:268 [Google Scholar]
[18]. Wedderburn LR, Rider LG, Juvenile Dermatomyositis: New Developments in pathogenesis, assessment and treatmentBest Pract Res Clin Rheumatol 2009 23(5):665-78. [Google Scholar]