Cystic Fibrosis (CF) is the second most common life-shortening monogenic disorder, after sickle cell anaemia with an incidence of 1 in 3700 [1]. It is an autosomal recessive disorder that presents as a multisystem disease with significant morbidity and mortality in all parts of the world caused due to an abnormal transport of chloride ions across the apical membranes of epithelial cells. This autosomal recessive genetic disorder is caused by mutations of the Cystic Fibrosis Trans membrane Conductance Regulator (CFTR) gene on chromosome 7q31.2, which encodes the CFTR chloride-ion channel that is an essential component of epithelial ion transport systems in many organs [2]. Due to wide range of native activities of CFTR protein in diverse organ systems, the spectrum of CF disease manifestations due to improper functioning of CFTR protein are wide. Classical CF is characterized by progressive lung disease, pancreatic dysfunction, elevated sweat chloride, meconium ileus, Congenital Bilateral Absence of Vas Deferens (CBAVD) and associated complications in untreated patients [3]. Pulmonary involvement occurs in 90% of patients surviving the neonatal period. Respiratory failure due to irreversible destruction of the lungs is the predominant cause of mortality in CF patients [4,5].
Nearly 2000 CFTR mutations have been identified so far, out of which only 127 CFTR mutations meet both clinical and functional criteria consistent with disease [6]. Although genetic analysis of CFTR gene by molecular techniques may be useful in confirming the diagnosis [7,8] but sweat test for quantitative analysis of sweat chlorides using the method of pilocarpine iontophoresis has been the gold standard for almost a half century for the diagnosis of CF [9]. Approximately in 98% patients with CF, sweat chloride value remains elevated [10,11]. The diagnosis of CF could easily be made in the majority of cases based on typical clinical features and abnormal sweat chloride values. Other ancillary tests include pancreatic function test which include fecal fat analysis and other parameters [12,13].
It is estimated that one in every 15,100 African Americans and one in every 13,500 Hispanics are diagnosed with CF each year, compared to one in every 2,500 Caucasians [14]. It has been suggested that CF is more severe in Asians [15,16]. CF was considered to be extremely rare in India and due to strong belief that CF does not occur in India, it is often not considered as a possibility even if the diagnosis is considered. However, published reports, reviews and comments indicate that CF is probably far more common in people of Indian origin than previously thought but is under diagnosed or missed in the majority of cases [17].
Only few studies have been undertaken from the valley of Kashmir on CF and sweat chloride analysis and in the absence of proper literature we cannot draw conclusion about magnitude of CF in Kashmir valley. The number of CF patients in Kashmir valley is very large compared to the expected values as large number of patients suspected for CF attend Sher-I-Kashmir Institute of Medical Sciences, GB pant and associated hospitals of Srinagar. In view of the large number of patients suspected for CF across the valley there was a dire need for establishment of sweat testing in the department of Clinical biochemistry SKIMS, Srinagar as it is the only tertiary care hospital in Kashmir region. Before the establishment of this routine diagnostic facility at SKIMS, Srinagar, sweat testing was done as a research parameter which was a discontinuous process and most of the patients had to go out of valley for getting a sweat test done resulting in wastage of money and resources.
The present prospective study aimed at filling this gap with the establishment of sweat chloride testing facility for the first time in Kashmir valley and further to elucidate the relationship of sweat chloride levels with clinical features and pattern of CF and hence to get an idea about the real scenario of this disease in this region.
Materials and Methods
The present study was a hospital based prospective study in patients suspected for CF conducted in the Department of Clinical Biochemistry in collaboration with Department of Paediatrics, Sher-I-Kashmir Institute of Medical Sciences (SKIMS), Soura for a period of one and a half years from Sept 2013-March 2015. The study was approved by the Institute Ethical Clearance Committee (SKIMS).
Inclusion and Exclusion Criteria
The patients of any age group or sex with one or more clinical features; recurrent Respiratory Tract Infection (RTI), history of CF in siblings, positive new born screening test, failure to thrive, wheezing, recurrent pneumonia, meconium ileus, chronic diarrhoea, Steatorrhoea, chronic cough and bronchiectasis, who reported to the Department of Paediatrics, SKIMS were subjected to sweat chloride test. Patients with abnormal karyotype and acute illness like Anorexia nervosa, hypothyroidism, Addison’s disease and electrolyte misbalance were excluded from the study.
Sample Size
A total of 182 patients, suspected for CF were included in the study who were referred from different hospitals of the state for effective outreach. Complete history was taken from all the patients before testing. Physical examination was done to check the presence of any co morbid conditions/risk factors. All the procedures were performed after taking the proper consent from the patients and their care takers.
Sweat Testing
The test was done by a validated method and equipment (iontophoresis unit) [18]. Procedure involves Sweat stimulation, Sweat collection and Quantitative sweat measurement for chloride. Sweat stimulation and Sweat collection is based on the Gibson and Cooks methodology involving pilocarpine iontophoresis [9]. The quantitative estimation of chloride was done by Schales and Schales method with some modifications in the concentration of mercuric nitrate and Sodium chloride [19]. In Infants (up to 6months of age) Cl-≤29 mmol/L was considered as normal; 30 to 59 mmol/L as intermediate and ≥60mmol/L indicative of CF. Beyond Infancy (Between 6 months to 39years of age) Cl- ≥39mmol/l was considered as normal; 40 to 59mmol/l as intermediate and ≥60mmol/l indicative of CF.A positive test i.e., sweat chloride concentration ≥60 mmol/l on two occasions in presence of suggestive clinical features was taken as a standard for the diagnosis of CF [20]. Patients with borderline values were put on follow-up and sweat testing was repeated after every 2months till final diagnosis. A negative test was repeated only under strong clinical suspicion.
CFTR Mutational Analysis
Corresponding blood samples with borderline sweat chloride levels were send to advanced centre for human genetics, SKIMS for CFTR mutational analysis. The samples were analysed for Δ508 Del, 3849 A>T, W1282X, 551, 541, 621+1 & R117 mutations.
Statistical Analysis
Univariate and multivariate binary logistic regression was used in our set of data. Descriptive statistics was also done for getting the mean, median and standard deviation of the data set. All the data was analysed using SPSS version 16.0 statistical software. Statistical Significance was taken when p<0.05.
Results
Of the 182 patients, males were 123 (67.5%) and 59 (32.5%) were females, with a male female ratio of 2:1. The mean age of the clinically suspected patients was 4.05±4.6 with a range of 0.2-23 years. The general characteristic of these182 high risk patients of cystic fibrosis is given in [Table/Fig-1].
Characteristics of high risk patients for sweat chloride analysis.
| S. No | Parameter | (n=182) |
|---|
| 1. | Sex | MaleFemale | 123(67.5)59(32.5) |
| 2. | Residence | RuralUrban | 152(83.5)31(16.5) |
| 3. | Referral Hospital | GB PantSKIMS | 105(57.6)77(42.4) |
| 4. | Age in years | ≤0.50.5-22-15>15 | 34(18.6)75(41.4)62(34.0)11(6.0) |
| 5. | RTI | AbsentPresent | 27(14.8)155(85.2) |
| 6. | Wheeze | AbsentPresent | 125(68.6)57(31.3) |
| 7. | Chronic Cough | AbsentPresent | 35(19.2)147(80.8) |
| 8. | Pneumonia | AbsentPresent | 153(84)29(16) |
| 9. | FTT | AbsentPresent | 124(70)29(16) |
| 10. | Diarrhoea | AbsentPresent | 137(75.2)45(24.8) |
| 11. | Bronchiectasis | AbsentPresent | 141(77.5)41(22.5) |
| 12. | History of CF in Siblings | AbsentPresent | 177(97.2)05(2.8) |
| 13. | Nasal Polyp | AbsentPresent | 174(95.6)08(4.4) |
| 14. | Product of CM | AbsentPresent | 141(77.5)41(22.5) |
| 15. | Meconium ileus | AbsentPresent | 176(96.7)06(3.3) |
| 16. | Digital Clubbing | AbsentPresent | 178(97.8)04(2.2) |
| 17. | Steatorrhea | AbsentPresent | 111(61)77(39) |
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
All the patients were subjected to pilocarpine iontophoresis for estimation of sweat chloride levels. It was made sure that the enrolled patients were clinically stable and fulfilling all the inclusion and exclusion criteria. The prerequisite age and weight of subjects for sweat testing was>2weeks and >3kg respectively. Out of 182 high risk patients 132 (72.5%) were having normal sweat chloride levels. Borderline and elevated sweat chloride levels were present in 9 (05%) and 41 (22.5%) subjects respectively. Pilocarpine Iontophoresis was repeated when inadequate sweat (<75mg) was obtained. Sweat testing was repeated in case of patients having normal and borderline sweat chloride levels but clinical picture highly suggestive of CF. Diagnosis of CF was not made on the basis of a single positive test result instead test was repeated in case of elevated sweat chloride levels. The district wise and hospital wise distribution of 50 patients having above normal sweat chloride levels (>39mmol/l) are given in [Table/Fig-2]. The mean age at diagnosis was 3.5 years with a range of 0.2-23 years. The mean sweat chloride values for positive and borderline cases were 94.7 and 48.5mmol/l respectively.
District wise distribution of positive cases.
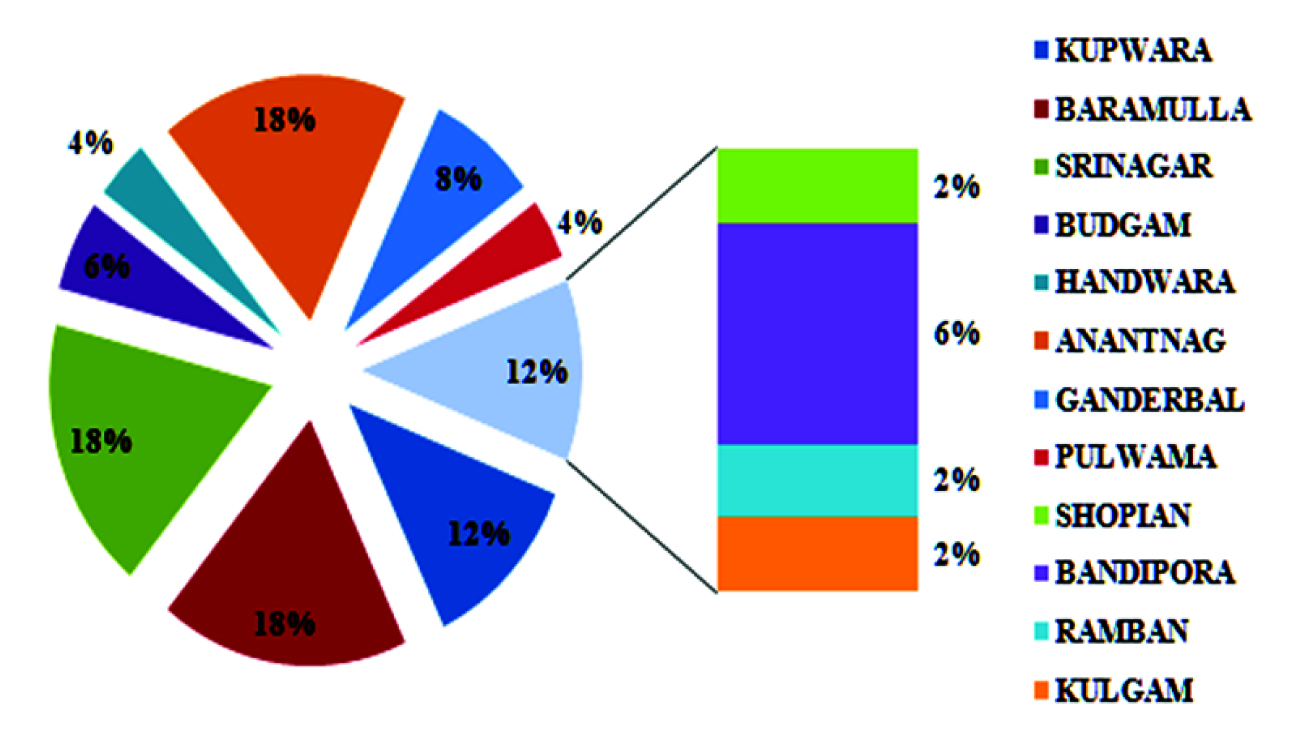
Most of the patients were having normal sweat chloride levels followed by elevated and borderline levels. There was a higher frequency of females (28.8%) with elevated sweat chloride levels than males (19.5%). Almost one fourth of the patients with RTI and chronic cough were having elevated sweat chloride levels. The other characteristics of high risk patients and their corresponding sweat chloride values are depicted in [Table/Fig-3].
Sweat chloride levels of patients with respect to their characteristics.
| S. No | Parameter | (n=182) | Sweat chloride levels (n=182) |
|---|
| Normal (N)n=132 (72.5%) | Borderline (B)n= 09 (05%) | Elevated (E)n=41 (22.5%) |
|---|
| 1. | Sex | MaleFemale | 123(67.5)59(32.5) | 95 (77.2)37 (62.7) | 04 (3.3)05 (8.5) | 24 (19.5)17 (28.8) |
| 2. | Residence | RuralUrban | 152(83.5)31(16.5) | 108(71.1)24(77.4) | 08(5.3)01(3.2) | 36(23.6)06(19.4) |
| 3. | Referral Hospital | OthersSKIMS | 105(57.6)77(42.4) | 84(80)48(62.3) | 03(2.9)06(7.8) | 18(17.1)23(29.9) |
| 4. | Age in years | ≤0.50.5-22-15>15 | 34(18.6)75(41.4)62(34.0)11(6.0) | 27(79.4)59(78.7)40(64.5)06(54.5) | 01(2.9)01(1.3)04(6.5)03(27.3) | 06(17.7)15(20)18(29.0)02(18.2) |
| 5. | RTI | AbsentPresent | 27(14.8)155(85.2) | 23 (85.2)109 (70.3) | 00 (00)09 (5.8) | 04 (14.8)37 (23.9) |
| 6. | Wheeze | AbsentPresent | 125(68.6)57(31.3) | 110 (88)22 (38.6) | 05 (04)04(7.1) | 10(08)31(54.4) |
| 7. | Chronic Cough | AbsentPresent | 35(19.2)147(80.8) | 28(80)104(70.7) | 01(2.8)08(5.5) | 06(17.2)35(23.8) |
| 8. | Pneumonia | AbsentPresent | 153(84)29(16) | 111(72.6)21(72.4) | 08(5.2)01(3.5) | 34(22.2)07(24.1) |
| 9. | FTT | AbsentPresent | 124(70)58(30) | 103(83.1)29(50) | 07(5.6)02(3.5) | 14(11.3)27(46.5) |
| 10. | Diarrhoea | AbsentPresent | 137(75.2)45(24.8) | 101(73.8)31(68.9) | 08(5.7)01(2.2) | 28(20.5)13(28.9) |
| 11. | Bronchiectasis | AbsentPresent | 141(77.5)41(22.5) | 108(76.6)24(58.5) | 05(3.5)04(9.8) | 28(19.9)13(31.7) |
| 12. | History of CF in Siblings | AbsentPresent | 177(97.2)05(2.8) | 132(74.6)00(00) | 09(5.0)00(0) | 36(20.4)05(100) |
| 13. | Nasal Polyp | AbsentPresent | 174(95.6)08(4.4) | 128(73.6)04(50) | 09(5.2)00(0) | 37(21.2)4(50) |
| 14. | Product of CM | AbsentPresent | 141(77.5)41(22.5) | 106(75.2)26(63.4) | 09(6.4)00(00) | 26(18.4)15(36.6) |
| 15. | Meconium ileus | AbsentPresent | 176(96.7)06(3.3) | 128(72.7)04(66.7) | 09(5.1)00(00) | 39(22.2)02(33.3) |
| 16. | Digital Clubbing | AbsentPresent | 178(97.8)04(2.2) | 132(74.2)00(00) | 09(5.1)00(00) | 37(20.7)04(100) |
| 17. | Steatorrhea | AbsentPresent | 111(61)77(39) | 97(87.4)35(45.5) | 02(1.8)07(9.1) | 12(10.8)35(45.4) |
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
In our study, when Normal sweat chloride levels were compared with borderline sweat chloride levels no significance was found for majority of the parameters except patients having Steatorrhoea were positively associated with borderline sweat chloride levels {p=0.003; OR = 9.7 (2.0-49.0)}. Overall Age was significantly associated with borderline sweat chloride levels with 03 out of 11 having >15years of age having borderline sweat chloride levels. [Table/Fig-4] shows the relation between various parameters and borderline sweat chloride levels of high risk patients. Elevated sweat chloride levels were significantly associated with many characteristics of high risk cases. Elevated sweat chloride levels were was significantly associated with wheeze {p = 0.0001; OR = 15.5 (6.6-36.0)}, FTT {p = 0.0001; OR = 6.8 (3.2-14.7)}, History of CF in Siblings{p = 0.0008; OR = 21.5 (2.5-84.0)}, Product of CM {p = 0.03; OR = 2.3 (1.1-5.0)}, Digital Clubbing {p = 0.003; OR = 17.5 (2.0-54.3)} and Steatorrhoea {p = 0.0001; OR = 8.1 (3.7-17.3)}. When elevated sweat chloride levels were compared with normal sweat chloride levels with respect to various parameters using multivariate analysis Wheeze, FTT and Steatorrhoea were found to be statistically significant. [Table/Fig-5&6] shows the association between normal and elevated sweat chloride levels with respect to the characteristics of high risk patients on univariate and multivariate analysis respectively. Among the 09 borderline cases 06 cases were positive for at least two CFTR gene mutations and rest of the 03 cases were not having any mutation in CFTR gene.
Association between normal and borderline sweat chloride levels with respect to the characteristics of high risk patients.
| S. No | Parameter | Sweat chloride levels | Odds ratio | p-value |
|---|
| Normal (N)n=132 | Borderline (B)n= 09 |
|---|
| 1. | Sex | MaleFemale | 9537 | 0405 | 3.2 (0.8-12.6) | 0.12 |
| 2. | Residence | RuralUrban | 10824 | 0801 | 0.6 (0.06-4.7) | 1.0 |
| 3. | Referral Hospital | GB PantSKIMS | 8448 | 0306 | 3.5 (0.8-14.6) | 0.08 |
| 4. | Age in years | ≤0.50.5-22-15>15 | 27594006 | 01010403 | - | 0.01 |
| 5. | RTI | AbsentPresent | 23109 | 0009 | 2.2 (0.26-17.8) | 0.7 |
| 6. | Wheeze | AbsentPresent | 11022 | 0504 | 4.0 (1.0-16.0) | 0.06 |
| 7. | Chronic Cough | AbsentPresent | 28104 | 0108 | 2.1 (0.25-18.0) | 0.7 |
| 8. | Pneumonia | AbsentPresent | 11121 | 0801 | 0.6 (0.07-5.5) | 1.0 |
| 9. | FTT | AbsentPresent | 10329 | 0702 | 1.0 (0.2-5.1) | 1.0 |
| 10. | Diarrhoea | AbsentPresent | 10131 | 0801 | 0.4 (0.05-3.4) | 0.5 |
| 11. | Bronchiectasis | AbsentPresent | 10824 | 0504 | 3.6 (0.9-14.4) | 0.07 |
| 12. | History of CF in Siblings | AbsentPresent | 13200 | 0900 | 13.3 (0.8-28.8) | 0.14 |
| 13. | Nasal Polyp | AbsentPresent | 12804 | 0900 | 3.2 (0.4-31.6) | 0.3 |
| 14. | Product of CM | AbsentPresent | 10626 | 0900 | 0.4 (0.5-3.2) | 0.5 |
| 15. | Meconium ileus | AbsentPresent | 12804 | 0900 | 2.5 (0.3-24.2) | 0.4 |
| 16. | Digital Clubbing | AbsentPresent | 13200 | 0900 | 13.3 (0.8-28.7) | 0.1 |
| 17. | Steatorrhoea | AbsentPresent | 9735 | 0207 | 9.7 (2.0-49.0) | 0.003 |
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
Association between normal and elevated sweat chloride levels with respect to the characteristics of high risk patients
| S. No | Parameter | Sweat chloride levels | Odds ratio | p-value |
|---|
| Normal (N)n=132 | Elevated (E)n=41 |
|---|
| 1. | Sex | MaleFemale | 9537 | 2417 | 1.8 (0.8-3.7) | 0.12 |
| 2. | Residence | RuralUrban | 10824 | 3606 | 0.7 (0.3-2.0) | 0.6 |
| 3. | Referral Hospital | GB PantSKIMS | 8448 | 1823 | 2.2 (1.1-4.5) | 0.03 |
| 4. | Age in years | ≤0.50.5-22-15>15 | 27594006 | 06151802 | - | 0.01 |
| 5. | RTI | AbsentPresent | 23109 | 0437 | 2.0 (0.6-6.0) | 0.32 |
| 6. | Wheeze | AbsentPresent | 11022 | 1031 | 15.5 (6.6-36.0) | 0.0001 |
| 7. | Chronic Cough | AbsentPresent | 28104 | 0635 | 1.6 (0.6-4.1) | 0.38 |
| 8. | Pneumonia | AbsentPresent | 11121 | 3407 | 1.1 (0.4-2.7) | 1.0 |
| 9. | FTT | AbsentPresent | 10329 | 1427 | 6.8 (3.2-14.7) | 0.0001 |
| 10. | Diarrhoea | AbsentPresent | 10131 | 2813 | 1.5 (0.7-3.2) | 0.28 |
| 11. | Bronchiectasis | AbsentPresent | 10824 | 2813 | 2.1 (0.9-4.6) | 0.08 |
| 12. | History of CF in Siblings | AbsentPresent | 13200 | 3605 | 21.5 (2.5-84.0) | 0.0008 |
| 13. | Nasal Polyp | AbsentPresent | 12804 | 3704 | 3.5 (0.8-14.5) | 0.09 |
| 14. | Product of CM | AbsentPresent | 10626 | 2615 | 2.3 (1.1-5.0) | 0.03 |
| 15. | Meconium ileus | AbsentPresent | 12804 | 3902 | 1.6 (0.3-9.3) | 0.1 |
| 16. | Digital Clubbing | AbsentPresent | 13200 | 3704 | 17.5 (2.0-54.3) | 0.003 |
| 17. | Steatorrhoea | AbsentPresent | 9735 | 1235 | 8.1 (3.7-17.3) | 0.0001 |
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
Parameters statistically significant on multivariate analysis between normal and elevated sweat chloride levels.
| S. No | Variable | Wald | OR (95% CI) | p-value |
|---|
| 1. | Wheeze AbsentWheeze Present | 18.86 | 0.05 (0.015-0.20) | 0.000 |
| 2. | FTT AbsentFTT present | 7.01 | 0.2 (0.06-0.6) | 0.008 |
| 3. | Steatorrhea AbsentSteatorrhea Present | 9.85 | 0.15 (0.04-0.5) | 0.000 |
FTT; Failure to thrive.
The patients diagnosed positive for CF were being treated and put on follow-up at our recently established CF clinic. The patients with normal sweat test were evaluated for other diseases in the department of paediatrics.
Discussion
CF is an inherited disease that causes the body to produce mucus that is extremely thick and sticky. The two organs most affected are the lungs and pancreas, where the thick mucus causes breathing and digestive problems. The disease was first described in an Indian patient in 1968 from the Post Graduate Institute of Medical Education and Research, Chandigarh [21]. Since then, the published data on Indian CF patients, however, has been very limited. In a study by Kabra et al., CF was detected in 3.5% of all children attending their paediatric chest clinic over a period of seven years [17]. The precise incidence of CF in India is still not known. From Kashmir, Tasaduq et al., did an observational study to describe the clinical profile, and delays in diagnosing CF patients. CF was diagnosed in 0.8% of enrolled patients with 6.5years as median age of presentation [22]. Masarat et al., screened 100 consecutive children patients of Kashmir valley presenting with one or more phenotypic features suggestive of CF by quantitative sweat chloride testing wherein positive sweat chloride (>60meq/l) was seen in 14 (14%) patients and 4 (4%) patients had equivocal (40-60meq/l) value on two different occasions [23].
In the present study we registered 182 suspected patients in one and a half years in Sher-I-Kashmir Institute of Medical Sciences, the only super speciality hospital of the valley. From the results of present study a precise incidence of CF cannot be measured but it can be said that there were patients from almost all the districts of Kashmir valley. Diagnosis of CF in our patients was based on the guidelines of CF Foundation in its Consensus Report in 2008 for diagnosis of CF [20], i.e., “Presence of characteristic phenotypic features” or “History of CF in siblings” or “A positive new-born screening test result” plus “An increased sweat chloride concentration” or “Identification of two CF-causing mutations” or “Demonstration of abnormal nasal epithelial ion transport”.
Sweat testing is a general term referring to the quantitative or qualitative analysis of sweat to determine electrolyte concentration, conductivity, or osmolality for the diagnosis of CF [10]. Although sweat chloride levels may be elevated in several conditions such as Anorexia nervosa, Hypothyroidism, Addison’s disease, Ectodermal dysplasia, Glycogen storage disease, oedema [22], the chances of false positive were very remote as patients were excluded from the above conditions and were having characteristic phenotypic features or History. Patients on mineralocorticoids, Malnutrition/Hypoproteinemia, Dehydration were excluded to avoid false negative results [24].
The facility for routine sweat testing was not available in any part of the Kashmir valley and the patients had to travel hundreds of kilometres to get sweat test done. We did a comprehensive sweat analysis for the first time in valley. Sweat testing was carried out in accordance with the guidelines given in a consensus report (CLSI, document C34-A3). Alternative procedures are no longer acceptable for the diagnosis of CF (Clinical and Laboratory Standards Institute –CLSI: A4 C3).
In our set of patients sweat chloride was elevated in 41 out of 182 patients (22.5%). Nine out of 182 (05%) were borderline for sweat chloride levels so the incidence of CF according to our study was quite high compared to other parts of India [25]. Further studies are needed to get the real scenario of CF in Kashmir valley. The average sweat chloride levels for positive cases were 94.7mmol/L with a minimum of 60mmol/L and maximum of 150mmol/L. The average sweat chloride levels for borderline cases were 48.5 mmol/L with a minimum of 40mmol/L and maximum of 55mmol/L. Females were mostly affected as per our study (24 of 41).
The average age at diagnosis was 3.5years ranging from 0.2-23years in our patients which although lower than 4.8years reported by a study done by Singh et al., 4.78years reported by Meenu et al., and 9.6years by Hamosh et al., [26,27], but much higher than the patients of USA where 71% of CF cases were diagnosed in the first year of life [28] suggesting the delay in diagnosis. This shows the low index of suspicion for the disease in our society and need of awareness about the occurrence of CF in Kashmiri children. Many studies suggest that CF is more severe in patients of Asian origin due to delayed diagnosis [10,15,26]. In western countries, diagnosis is delayed due to absence of typical clinical features or milder illnesses while delayed diagnosis in North Indian children is due to lack of awareness and diagnostic facilities. Early mortality due to severe disease in neonatal or infantile period is one of the main reasons for masking the high incidence of CF in this region. An earlier diagnosis and therefore intervention is associated with improved growth, reduced morbidity and reduced therapy.
Although the clinical profile of our CF patients is similar to that classically described [10,25,29] pulmonary disorders including recurrent respiratory tract infections, wheeze, chronic cough, Pneumonia, Bronchiectasis were common manifestation of CF patients with positive sweat chloride levels. Wheeze was present in 75.6% (31 of 41) of patients diagnosed with CF and was significantly associated with positive sweat chloride levels (p<0.05) which is slightly lower than 80.6% reported by Uzma et al., and Naguib et al., reported abnormal chest examination in 92% of CF patients [30,31]. In North American CF registry, 48.8% had respiratory symptoms [1] (Cystic fibrosis Foundation Patient Registry (CFFP) registry, 2014). Bronchiectasis was mainly due to pseudomonas colonization which was evident by CT chest of CF patients showing mild bronchial wall thickening and mucous plugging which again reflects the time of lag for Pseudomonas aurignosa to colonize and hence leading to delay in diagnosis.
Almost 70% (27 of 41) of CF patients had FTT and was significantly associated with elevated sweat chloride levels (p<0.05) which is less as compared to the studies done by Naguib et al., and Al-Mobaireek et al., where 100% of CF patients had FTT probably due to inclusion of high risk patients in these studies [31,32] but greater than the frequency of 32.0% reported by CFFP Registry, 2013 [1]. Uzma et al., reported FTT in 81.9% of CF patients [30]. Digital clubbing was present in 10% (04 of 41) of CF patients having elevated sweat chloride levels which is lower than the frequency of 80% and 25% reported by Kabra et al., and Naguib et al., respectively [10,31]. Although the frequency of digital clubbing is low in our study population but is absent in non CF patients, hence significantly associated with elevated sweat chloride levels (p< 0.05).
Pancreatic insufficiency leading to diarrhoea, Steatorrhoea etc. was common in our set of patients with a significant association with Steatorrhoea. Steatorrhoea was present in 85.3% (35 of 41) of CF patients. Diarrhoea was present in 31.7% (13 of 41) of patients in our study which is comparable to 43.6% reported by CFFP Registry, 2013 [1] but very less than the frequency of 100% reported by Al-Mobaireek et al., [32]. During our study, the cases presenting pancreatic abnormalities were observed to possess a higher age group indicating that the damage of pancreas in utero occurs progressively and the patients can present with symptomatic pancreatic abnormalities as the initial manifestations of CF in adulthood [33].
Almost 12.1% (05 of 41) of patients had history of CF in siblings which was statistically significant (p<0.05). The study revealed high rate of consanguinity (36.5%; 15 of 41) in CF group with significant association with positive sweat chloride status (p<0.05). This reflects the overall high incidence of consanguinity in a closed society like ours and thus increasing the probability of recessive disorders like CF. No other study has correlated the sweat chloride levels with different characteristics of high risk patients as comprehensively as in the present study.
Our study has reported 5% (9 of 182) borderline cases (sweat chloride levels 40-59mmol/L). In these patients the diagnosis remains in doubt and according to the CFF guidelines the patients should be evaluated for CFTR mutations to have a clear picture of the disease. A panel of 7 mutations in CFTR gene was done in these 9 patients in Advance Centre for Human Genetics, Sher-I-Kashmir Institute of Medical sciences. The patients were evaluated for Δ508 Del, 3849 A>T, W1282X, 551, 541, 621+1 and R117 mutations. Out of 9 patients 6 were having at least two mutations in the CFTR gene thus satisfying the criteria for cystic fibrosis. The rest 3 patients were negative for the mutations and the diagnosis was not clear in those three patients. Such cases are now labelled as cystic fibrosis trans membrane Conductance Regulator related Metabolic Syndrome (CRMS) which is proposed to describe infants who have sweat chloride values ranging from 40-59mmol/L and up to two CFTR mutations, at least one of which is not clearly categorized as a “CF-causing mutation”, thus they do not meet CF Foundation criteria for the diagnosis of CF [34]. These patients have a milder clinical course and are more likely to have pulmonary insufficiency [35,36], but have pancreatic sufficiency. These patients may have a different prognosis from patients with “Classic CF”. According to CFFP Registry 2013 [1] there is a panel of 25 mutations to be done for accessing the CFTR status. The reason for the negative mutational status of 3 patients might be due to the fact that only seven mutations were done in Advance Centre for Human Genetics, Sher-I-Kashmir Institute of Medical sciences (SKIMS) raising the possibility weather CFTR mutations were really absent or it was missed in these patients.
Conclusion
Genetic mutation profile being heterozygous is unlikely to become diagnostic test; hence, sweat testing remains a gold standard for diagnosis of CF patients. Although we need to enhance the panel of CFTR mutations for accurate diagnosis of CF but neonatal screening program for CF may improve the diagnosis and answer the magnitude of problem as well. CF does occur in Indian children especially Kashmir valley, however, precise magnitude of problem is not known. The clinical features are classical. Diagnosis is often delayed and the disease is advanced in most patients at the time of diagnosis. Apart from creating awareness about CF in doctors it is also necessary to develop diagnostic facilities and service centres for diagnosis and treatment of CF patients. There is need to develop model for management of these patients based on locally available resources.
Author’s Contribution
MAR and MSK: First two authors have contributed equally (Have done the major experimental work, Provided the basic idea;standardized the Pilocarpine iontophoresis along with titration; drafted and edited the manuscript). SAM: Provided the experimental support. AHR: Recruited and referred patients to the lab. MJM: Provided the experimental support. JIB: Recruited and referred patients to the lab. Treated and followed up the patients in CF clinic. SM: Provided the Lab and Basic facilities for the whole study.
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
RTI; Respiratory tract infection, FTT; Failure to thrive, CM; Consanguineous marriage
FTT; Failure to thrive.