Cardiovascular Abnormalities in Egyptian Children with Mucopolysaccharidoses
Laila Selim1, Nehal Abdelhamid2, Emad Salama3, Amera Elbadawy4, Iman Gamaleldin5, Mohamed Abdelmoneim6, Abeer Selim7
1 Professor, Department of Paediatrics, Faculty of Medicine, Cairo University, Cairo, Egypt.
2 Researcher, Department of Paediatrics, National Research Center, Cairo, Egypt.
3 Professor, Department of Paediatrics, National Research Center, Cairo, Egypt.
4 Consultant, Department of Paediatrics, Faculty of Medicine, Cairo University, Cairo, Egypt.
5 Research Assistant, Department of Paediatrics, Faculty of Medicine, Cairo University, Cairo, Egypt.
6 Lecturer, Department of Clinical Pathology, Faculty of Medicine, Cairo University, Cairo, Egypt.
7 Researcher, Department of Paediatrics, National Research Center, Cairo, Egypt.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Nehal Abdelhamid, National Research Center, El Buhouth St, Dokki, Cairo-12311, Egypt.
E-mail: ibra_n2005@yahoo.com
Introduction
The Mucopolysaccharidoses (MPS) are rare inherited metabolic disorders. They are characterized by the progressive systemic deposition of Glycosaminoglycans (GAGs). GAGs accumulate in the myocardium and the cardiac valves. Enzyme Replacement Therapy (ERT) is available for MPS I, II, and VI. However, ERT does not appear to improve cardiac valve disease in patients with valve disease present at the start of ERT.
Aim
To evaluate the cardiac involvement in Egyptian children with MPS.
Materials and Methods
Echocardiograms (ECG) were done for 34 patients. Both quantitative and qualitative Glycosaminoglycans (GAGs) in urine and enzyme assay confirmed the diagnosis. Mitral, tricuspid and aortic valves were evaluated for increased thickness, regurgitation and/or stenosis, left ventricular chamber dimensions, septal and posterior wall thicknesses.
Results
The patients’ age ranged from 0.9-16 years (median age 4 years). They included 19 cases of MPS I (55.9%), 3 cases of MPS II (8.8%), 2 cases of MPS III (5.9%), 6 cases of MPS IV (17.6%) and 4 cases of MPS VI (11.8%). Heart murmur was heard in 9 of the participants (9/34) (26%). However, 15 patients (15/34) (44%) revealed cardiac lesions on ECG examinations. Mitral regurge (47%), followed by pulmonary hypertension (40%), were the most frequent findings.
Conclusion
The absence of Cardiac murmurs does not exclude the heart involvement. Cardiac valve dysfunction may not be reversible. Regular ECG should be routinely warranted in children with MPS and early ERT are recommended.
Echocardiography, Enzyme replacement theory, Paediatrics
Introduction
Mucopolysaccaridoses (MPS) are rare metabolic disorders that are inherited in an autosomal recessive pattern, and demonstrate cardiovascular involvement [1]. Depending on the type of enzyme deficiency, the catabolism of Dermatan Sulfate (DS), Heparan Sulfate (HS), Keratin Sulfate (KS), chondroitin sulfate or hyaluronan may be blocked. Lysosomal accumulation of GAGs molecules results in cells, tissues and organ dysfunction, including the heart [2,3]. Children with MPS display clinical symptoms that include skeletal abnormalities, CNS deterioration, cardiac dysfunction, corneal clouding, loss of hearing, enlarged abdominal organs, and obstructive airway disease [4]. All MPS types have an autosomal recessive inheritance except type II, which has an X linked inheritance. Despite the biochemical and genetic heterogeneity, different MPS types have similar clinical presentation [5]. Urinary Glycosaminoglycans (GAGs) levels and enzyme activity assays are required to confirm variable MPS types. Measurement of enzyme activities in leukocytes or cultured fibroblasts is currently the gold standard for diagnosing a specific MPS type(s) [6,7]. The skeletal abnormalities are collectively referred to as dysostosis multiplex. The characteristic radiographic findings include paddle-shaped ribs, thick clavicles, odontoid hypoplasia, platyspondyly, wedge-shaped vertebral bodies with anterior breaking, dorsal kyphosis, lumbar gibbus, wide disc spaces and spinal canal stenosis. These skeletal changes result in restricted mobility, arrest of growth in childhood and short stature [8].
Deposition of GAGs in the heart leads to cardiomyopathy or cardiac valve disease [9,10]. Nevertheless, development of coronary artery disease and sub-clinical atherosclerosis in children with severe Mucopolysaccharidosis (MRS) type I and cardiomyopathy has been reported [11,12]. Cardiorespiratory disease tends to progress with age and is the most common cause of death. With the development of Enzyme Replacement Therapy (ERT) for many of these disorders, the results of therapies will rely heavily upon early detection and treatment, before irreversible damages. Long-term ERT is beneficial for the myocardium, but the cardiac valves involvement appears irrevesible [13,14].
The present study was conducted to evaluate the cardiac involvement in Egyptian children with MPS.
Materials and Methods
This was a cross-sectional study of 34 pediatric patients with MPS who attended the neurometabolic clinic at Cairo University Children Hospital, from January 2013 to June 2014. The study was approved by the Institutional Ethics Committee. The study was performed according to the principles of the Declaration of Helsinki (2008). Informed consent was obtained from all the parents. The mean age ±standard deviation of the participants was 5.7 ± 3.77 years and number of male patients was 23. The measurement of enzyme activities was done to confirm different types of MPS. Enzymatic assays were carried out according to the methods described by Kresse et al., [15].
All patients were subjected to thorough medical history assessments that included family history and a three-generation pedigree analysis of parental consanguinity and any similarly affected family members. Full clinical examination was done to detect features of MPS disorders.
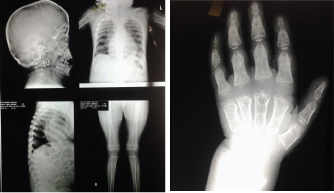
Skeletal survey was conducted for all cases to detect evidence of skeletal abnormalities [Table/Fig-1a,b]. Quantitative and electrophoretic analysis of urinary (GAGs) as well as enzymatic analysis for types of MPS was done to measure the activities of α-L-iduronidase, iduronate-2-sulfatase, α-N-acetylglucosaminidase, galactosamine-6-sulfatase, arylsulfatase B in the White Blood Cells [Table/Fig-2].
(a) Skeletal survey of a five-year-old girl with type VI. Enlarged cranium-J shaped sella turcica-enlarged heart-wide ribs-dorsolumber gibbus at D12/L1-anterior breaking of vertebral bodies. (b) The same patient’s hand- wide metacarpal, proximal and middle phalanges-pointed proximal ends of the second to fifth phalanges.
Investigations to confirm different types of MPS.
| MPS I Hurler(N = 19) | MPS II Hunter(N = 3) | MPS III Sanfilippo(N = 2) | MPS IV Morquio(N = 6) | MPS VI Maroteax-Lamy(N = 4) |
|---|
| Dysostosis multiplex | 18(95%) | 3(100%) | 2(100%) | 6(100%) | 3(75%) |
| GAG in Urine | DS,HS | DS,HS | HS | KS | DS |
| Enzyme assay | alpha – L-iduronidase | iduronate 2 sulphatase | Alpha-N-acetylglucos-aminidase | N acetyl galactosamine-6-sulfatase | aryl-sulphatase |
GAG: glycosaminglycan, DS;Dermatan sulphate, HS;Heparan sulphate, KS;Keratan sulphate.
Electrocardiogram (ECG) was performed for all cases in the supine, left lateral position using General Electric (GE, Vivid-5) system with probe 3 or 5 MHz (multi-frequency transducer) according to the age of patient, having tissue velocity imaging capabilities. The electrocardiography was connected to time the cardiac cycle events. The examination consisted of M-mode, two-dimensional, pulsed-wave and colour Doppler blood flow velocity assessments of the cardiac valves.
Other investigations, such as Magnetic Resonance Imaging (MRI) of brain and spine was done. Auditory brain stem evoked potential was obtained from those MPS patients with hearing problems (otitis media) and median nerve conduction velocity was done to diagnose cases of carpal tunnel syndrome.
Statistical Analysis
Statistical Package for Social Science (SPSS) program version 17 was used for data analysis. Data was described in terms of mean ± standard deviation (±SD), or frequencies (number of cases) and percentages when appropriate.
Results
The study was conducted on 34 patients, aged 0.9–16 years; the mean age (±standard deviation) of the participants was 5.7 ± 3.77 years, with 23 male and 11 female. There were 19 cases of MPS I (55.9%), 3 cases of MPS II (8.8%), 2 cases of MPS III (5.9%), 6 cases of MPS IV (17.6%) and 4 cases of MPS VI (11.8%). A total of 13 (38%) patients have positive family history of similar condition. Coarse facial features were present in all cases of Hunter and Sanfilippo Syndrome and were the most common finding in Hurler cases (70%). Corneal clouding was seen in Hurler and Hunter patients while absent in Sanfilippo, Morquio and Maroteaux-Lamy patients. Heart Murmur was heard in nine (9/34) cases (26%), with higher prevalence in Maroteaux-Lamy patients (50%). Hyperactivity was apparent in Sanfilippo cases. Median nerve conduction velocity showed bilateral median nerve neuropathy of axonal nature, in 6 patients (17.6%). It ranges between moderate to severe degrees. The findings of clinical characteristics are represented in [Table/Fig-3].
Frequency of the clinical findings of individuals with MPS.
| Hurler(N = 19) | Hunter(N =3) | Sanfilippo(N = 2) | Morquio(N = 6) | Maroteaux-Lamy(N = 4) |
|---|
| Coarse facial features | 19(100%) | 3(100%) | 2(100%) | 4(66.6%) | 4(100%) |
| Macrocephaly | 13(68%) | 1(33%) | 2(100%) | 4(66.6%) | - |
| Corneal clouding | 10(53%) | 1(33%) | - | - | 3(75%) |
| Chronic nasal congestion / snoring/Adenoids | 3(16%) | 2(66%) | - | - | 3(75%) |
| Heart murmur | 4(21%) | 1(33%) | - | 1(16.6%) | 2(50%) |
| inguinal and/or umbilical hernia | 10(53%) | 2(66%) | - | 1(16.6%) | 1(25%) |
| liver and spleen enlargement | 5(26%) | 2(66%) | 1(50%) | - | 2(50%) |
| Short stature | 10(53%) | 2(66%) | 1(50%) | 3(50%) | 2(50%) |
| Kyphosis | 2(10.5%) | 1(33%) | - | 2(33%) | - |
| Genu valgus | - | - | - | 2(33%) | - |
| Hearing loss | 1(5%) | - | - | 1(16.6%) | - |
| Hyperactivity | - | - | 2(100%) | - | - |
| Carpal tunnel syndrome | 2(10.5%) | 2(66%) | - | 2(33%) | |
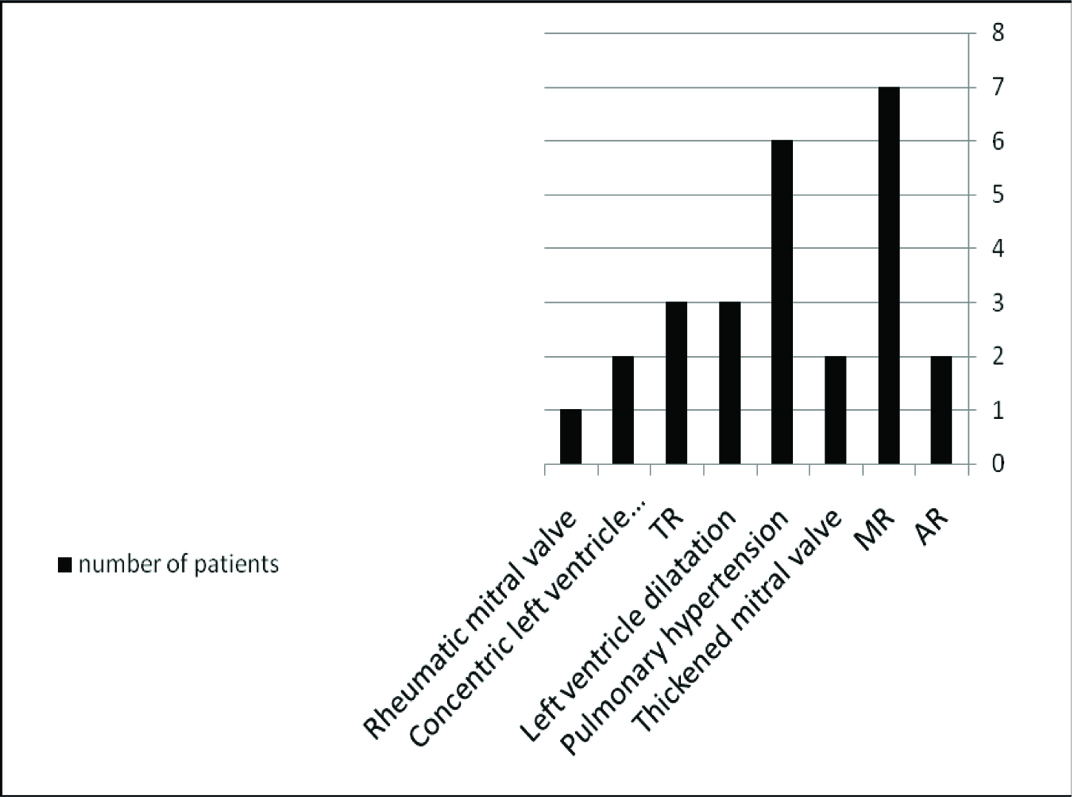
ECG was performed in all patients. Cardiac abnormalities were detected in 15 patients (44%) such as mitral valve lesions, pulmonary hypertension, concentric left ventricle hypertrophy/dilatation, tricuspid regurge and/or aortic regurge. Mitral regurge (47%) followed by pulmonary hypertension (40%) were the most common findings among MPS patients [Table/Fig-4]. We encountered 3 patients with MPS I in whom the predominating heart problems were infiltration of the valvular cusps, ventricular hypertrophy and/or dilatation leading to mitral valve insufficiency and pulmonary hypertension. Cardiac lesions in various types of MPS are shown in [Table/Fig-5]. ECG showed ST segment depression-abnormality and negative T-wave, in cases of concentric Left ventricle hypertrophy.
Echocardiographic findings in MPS Patients.
TR; Tricuspid Regurge, MR; Mitral Regurge, AR; Aortic regurge
Echocardiographic findings in various types of MPS.
| Types of MPS | Thickened mitral valve | Rheumatic mitral valve | TR | AR | MR | Lt ventricular dilation | Concentric Lt ventricular hypertrophy | Pulmonary hypertension |
|---|
| Hurler (N = 19) | 1 | - | 1 | - | 3 | 2 | 1 | 3 |
| Hunter (N = 3) | - | - | - | - | 2 | 1 | - | - |
| Sanfilippo(N = 2) | - | - | 1 | - | - | - | - | - |
| Morquio (N = 6) | 1 | 1 | 1 | - | 1 | - | - | 2 |
| Maroteaux Lamy (N = 4) | - | - | - | 2 | 1 | - | 1 | 1 |
TR; Tricuspid Regurge, MR; Mitral Regurge, AR; Aortic Regurge
One patient with Hunter disease died at the age of 10 years. His echo showed mitral regurge and dilated left ventricle. His death was thought to be related to the cardiac abnormalities.
MRI of brain and spine, were available in 31 cases [Table/Fig-6]. Out of 31, 24 cases (77%) have abnormal findings. Demyelination, incomplete myelination and dysmyelination are described in 11/31 patients (35%). Abnormal dorsolumbar vertebra was described in 2(6%) cases, while four patients (13%) had venticulomegaly; one of whom had ventriculoperitoneal shunt operation.
MRI finding in MPS patients.
| MRI findings | Number (%) |
|---|
| Progressive narrowing of foramen magnum | 4(13%) |
| Demylination | 7(22%) |
| Incomplete myelination | 3(10%) |
| Dysmyelination | 1(3%) |
| Bilateral symmetrical cortical atrophic changes | 3(10%) |
| Diffuse signal changes of periventricular white matter | 4(13%) |
| Moderate hydrochephalus with CSF permeation, cribriform appearance of periventricular white matter thalami and body of corpus callosum(dilated virchow Robin spaces-bilateral temporal arachnoid cyst | 1(3%) |
| Macrocranium with deformed craniocervical junction and cystic colossal changes | 1(3%) |
| Abnormal dorsolumbar vertebra | 2(6%) |
| Venticulomegaly | 4(13%) |
Discussion
Cardiac abnormalities have been observed in patients with MPS of any types. However, an early diagnosis is difficult due to joint stiffness, respiratory system involvement or skeletal malformations that limit exercise capacity and mask the underlining heart failure [16]. The most frequent MPS type in Egyptian children was type I (56%). The anatomical changes of cardiovascular systems were common in all types of MPS patients. In another study, among Taiwanese children, the most common type was type II (43%) [17]. In this study, the heart murmur was heard in 26% of our patients with MPS. Mitral regurgitation was the most common echo finding. Rigante and Signi found clinical evidence in most cases that have moderate-to-severe somatic disease [9]. Thickening of the valve leaflets, commonly leads to mitral and aortic regurgitation rather than stenosis [3].
It has been suggested that, cardiomyopathy and valve defects occur as GAGs accumulate in the myocardium, spongiosa of cardiac valves, and myointima of coronary arteries [5]. Histologically, cytoplasmic vacuolation may be observed in cells of mitral valves, in coronary arteries and in the aorta; electron-microscopically, the same cardiomyocytes may appear enlarged or packed with electron-lucent cytoplasmic vacuoles, which are stored undegraded GAGs [17]. The deformed valves showed mitral or aortic leaflets thickening and calcification, and may lead to various severities of stenosis or regurgitation. Pulmonary hypertension is the second echo finding in our patients. There was a strong association between the accumulation of dermatan sulphate and the presence of valvular dysfunction, and pulmonary hypertension [18]. Rheumatic mitral valve was diagnosed in one patient however, these findings could be attributed to infiltration of polysaccharides or mucosubstances instead of rheumatic involvement [10]. Moreover, multi-system disease involving the dermatological, musculoskeletal, cardiac and gastrointestinal system could suggest storage disease [19]. Cardiovascular abnormalities among patients with MPS III were rare and less severe than among other groups [20]. Similar to our study, one of those has mild tricuspid regurge.
Valvular deformation and thickening, thick interventricular septum and diastolic dysfunction were found to worsen with age [17]. Insofar, ECG and Echo should be done regularly during follow-up of MPS patients [21]. Valvular lesions causing valvular stenosis or insufficiency are an indication for infectious endocarditis prophylaxis before surgical and dental treatment [22]. Valvular disease, myocardial thickening, systemic and pulmonary hypertension [18] or narrowing of the coronary arteries with ischemia, all contribute to instances of sudden cardiovascular collapse [9].
Treatment options for MPS include, either haematopoietic stem cell transplantation, which is useful for selected patients or recombinant I.V enzyme replacement therapy. ERT is available for MPS I, II, VI [6], and recently for type IV [23]. ERT may alter the course of ventricular hypertrophy but cardiac valve pathology is usually unresponsive or, at best, stabilized [24]. In Taiwanese study of patients with MPS VI, ERT reduced urinary GAG and had positive effects on cardiac hypertrophy and diastolic dysfunction [25]. Early ERT may prevent valve disease, a fact that emphasizes the importance of early diagnosis and treatment in MPS [3].
Conclusion
Echocardiography is a key diagnostic technique for evaluation of valves, ventricular dimensions and function; even among asymptomatic patients. Serial ECG in patients with MPS are recommended to assess ventricular function and the progression of cardiac abnormalities. Prompt diagnosis and treatment are mandatory before irreversible damage.
GAG: glycosaminglycan, DS;Dermatan sulphate, HS;Heparan sulphate, KS;Keratan sulphate.
TR; Tricuspid Regurge, MR; Mitral Regurge, AR; Aortic Regurge
[1]. Goksel OS, El H, Tireli E, Dayioglu E, Combined aortic and mitral valve replacement in a child with mucopolysaccharidosis type I: a case reportJ Heart Valve Dis 2009 18:214-16. [Google Scholar]
[2]. Brands MMMG, Frohn-Mulder IM, Hagemans MLC, Hop WCJ, Oussoren E, Helbing WA, Mucopolysaccharidosis: cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VIJ Inherit Metab Dis 2013 36:227-34. [Google Scholar]
[3]. Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and managementJ Inherit Metab Dis 2011 34:1183-97. [Google Scholar]
[4]. Jackson M, Derrick Roberts A, Martin E, Rout-Pitt N, Gronthos S, Byers S, Mucopolysaccharidosis enzyme production by bone marrow and dental pulp derived human mesenchymal stem cellsMol Genet Metab 2015 114:584-93. [Google Scholar]
[5]. Cobos PN, Steglich C, Santer R, Lukacs Z, Dried blood spots allow targeted screening to diagnose mucopolysaccharidosis and mucolipidosisJIMD Rep 2015 15:123-32. [Google Scholar]
[6]. Muenzer J, Overview of the mucopolysaccharidosesRheumatology 2011 50:4-12. [Google Scholar]
[7]. Parkinson-Lawrence EJ, Muller VJ, Hopwood JJ, Brooks DA, N-acetylgalactosamine-6-sulfatase protein detection in MPS IVA patient and unaffected control samplesClin Chim Acta 2007 377:88-91. [Google Scholar]
[8]. Clarke LA, Hollak CE, The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disordersBest Pract Res Clin Endocrinol Metab 2015 29:219-35. [Google Scholar]
[9]. Rigante D, Segni G, Cardiac structural involvement in mucopolysaccharidosesCardiology 2002 98:18-20. [Google Scholar]
[10]. Mettananda DS, Fernando AD, Child with Morquio syndrome and mixed mitral valve diseaseCeylon Med J 2008 53:24-25. [Google Scholar]
[11]. van den Broek L, Backx AP, Coolen H, Wijburg FA, Wevers R, Morava E, Fatal coronary artery disease in an infant with severe mucopolysaccharidosis type IPediatrics 2011 127:e1343-46. [Google Scholar]
[12]. Wang RY, Braunlin EA, Rudser KD, Dengel DR, Metzig AM, Covault KK, Carotid intima-media thickness is increased in patients with treated mucopolysaccharidosis types I and II, and correlates with arterial stiffnessMol Genet Metab 2014 111:128-32. [Google Scholar]
[13]. Braunlin EA, Berry JM, Whitley CB, Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type IAm J Cardiol 2006 98:416-18. [Google Scholar]
[14]. Muenzer J, Early initiation of enzyme replacement therapy for the mucopolysaccharidosesMol Genet Metab 2014 111:63-72. [Google Scholar]
[15]. Kresse H, von Figura K, Klein U, Glössl J, Paschke E, Pohlmann R, Enzymic diagnosis of the genetic mucopolysaccharide storage disordersMethods Enzymol 1982 83:559-72. [Google Scholar]
[16]. Golda A, Jurecka A, Opoka-Winiarska V, Tylki-Szymańska A, Mucopolysaccharidosis type VI: a cardiologist’s guide to diagnosis and treatmentInt J Cardiol 2013 15:1-10. [Google Scholar]
[17]. Lin SM, Lin HY, Chuang CK, Lin SP, Chen MR, Cardiovascular abnormalities in Taiwanese patients with mucopolysaccharidosisMol Genet and Metab 2014 111:493-98. [Google Scholar]
[18]. Leal GN, De Paula AC, Leone C, Kim CA, Echocardiographic study of paediatric patients with mucopolysaccharidosisCardiol Young 2010 20:254-61. [Google Scholar]
[19]. Madireddi J, Sarada P, Shetty RK, Prabhu M, Girish KM, Hunter syndrome with its typical heart: a close mimic to rheumatic heartBMJ Case Rep 2015 7 [Google Scholar]
[20]. Gross DM, Williams JC, Caprioli C, Dominguez B, Howell RR, Echocardiographic abnormalities in the mucopolysaccharide storage diseasesAm J Cardiol 1988 61:170-76. [Google Scholar]
[21]. Tian Z, Guo LL, Meng Y, Zhang SY, Zhu WL, Cardiac involvement of the type I mucopolysaccharidosisZhonghua Nei Ke Za Zhi 2013 52:197-99. [Google Scholar]
[22]. Dangel JH, Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders: clinical and echocardiographic findings in 64 patientsEur J Pediatr 1998 157:534-38. [Google Scholar]
[23]. Lyseng-Williamson KA, Elosulfase Alfa: a review of its use in patients with mucopolysaccharidosis type IVA (Morquio A syndrome)Bio Drugs 2014 28:465-75. [Google Scholar]
[24]. Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 yearsMol Genet Metab 2007 90:171-80. [Google Scholar]
[25]. Lin HY, Chuang CK, Wang CH, Chien YH, Wang YM, Tsai FJ, Long-term galsulfase enzyme replacement therapy in Taiwanese mucopolysaccharidosis VI patients: A case seriesMol Genet Metab Rep 2016 7:63-69. [Google Scholar]