Cardiofaciocutaneous syndrome or CFC syndrome is a rare genetic disorder first described in 1986. It is one of the RASopathies involving multiple organs particularly the heart, skin and face affecting males and females equally. The phenotypic features overlap with 2 other conditions, the Noonan and Costello syndrome. We report on a 22-month-old boy with CFC syndrome presenting with typical craniofacial appearance, heart defects, ectodermal abnormalities, growth failure and developmental delay. Estimated population of affected individuals worldwide is a few hundreds.
Costello syndrome, Noonan syndrome, RASopathies
Case Report
A 22-month-old boy born out of non-consanguineous marriage presented to us with complaints of developmental delay (not being able to stand or walk at age of 22 month). The antenatal history was unremarkable except for polyhydramnios. Birth and perinatal history were uneventful. Baby weighed 9kg (below 2nd percentile) on admission, with a length and head circumference of 78cm (below 2nd percentile) and 51cm (more than 98th percentile) respectively.
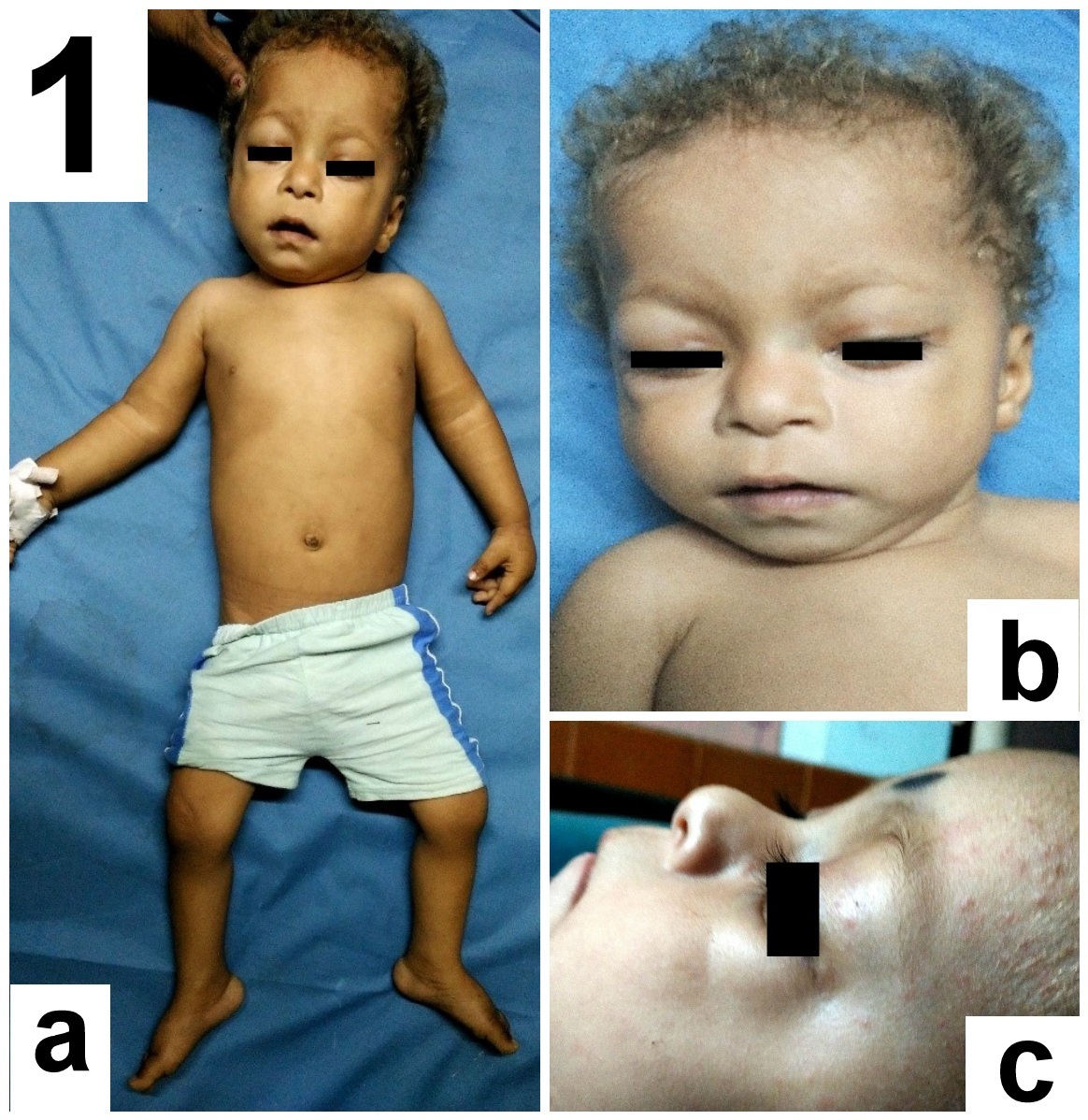
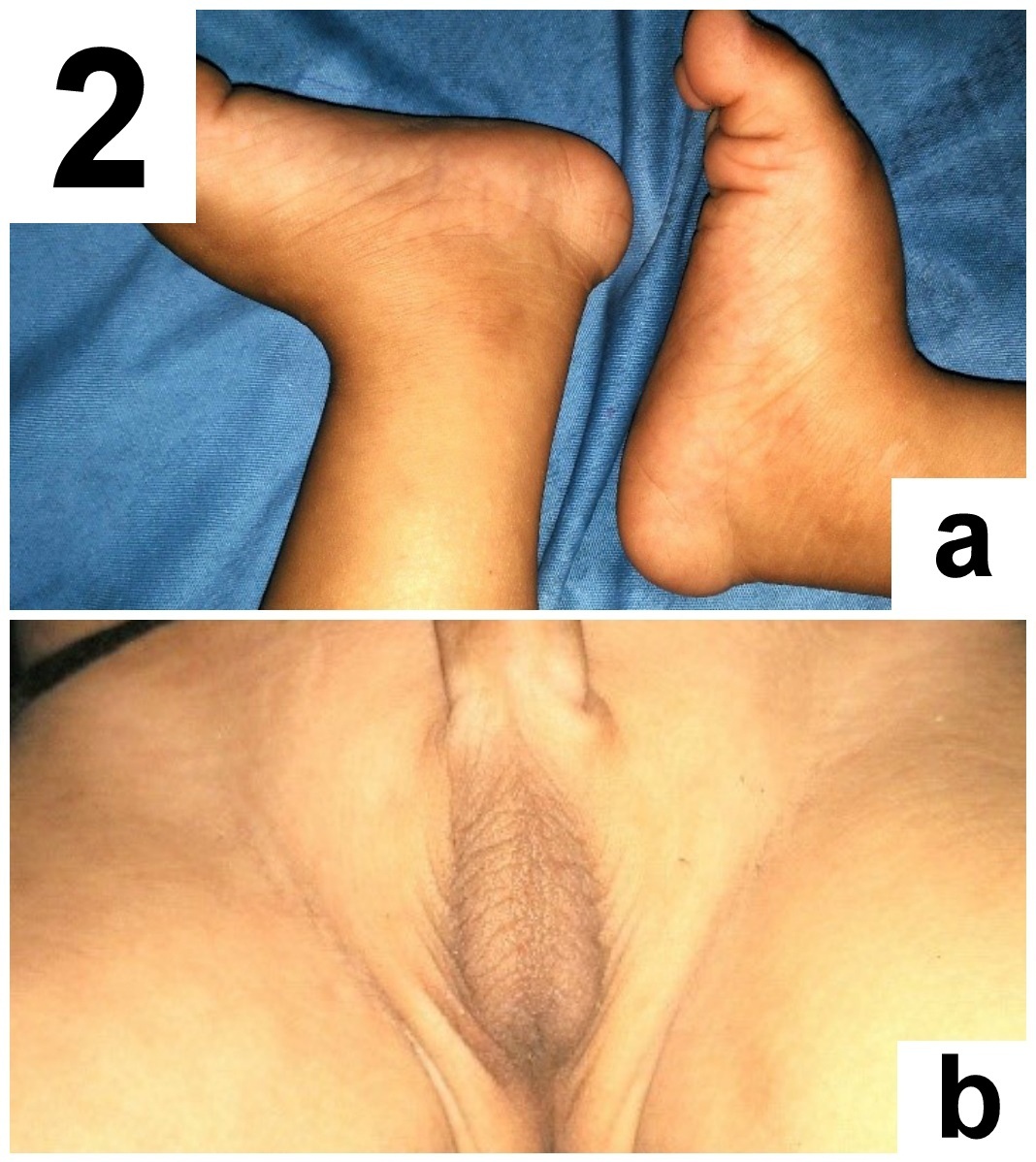
Detailed head to foot examination revealed relative macrocephaly, characteristic coarse facies having sparse curly hair, sparse eyebrows, low set ears, hypoplasia of supraorbital ridges, ocular hypertelorism, short nose with depressed nasal bridge, ptosis, deep philthrum [Table/Fig-1a-c]. Skin was dry with dystrophic nails. There was rocker bottom deformity on both the feet [Table/Fig-2a]. Genital examination revealed bilateral cryptochordism [Table/Fig-2b] with both testes being palpable in the inguinal canal. Ophthalmological examination revealed bilateral incomplete iris coloboma, grade 2. The child had a history of febrile seizure at 9 months of age. Global developmental delay was present. Neurological examination revealed hypotonia in all four limbs, with normal power and reflexes. Auscultation of chest revealed an ejection systolic murmur in the pulmonary area. Magnetic Resonance Imaging (MRI) brain was normal without any atrophy.
(a). Showing overall appearance with characteristic facial features, dry skin and bilateral rockerbottom feet. (b) Characteristic coarse facies showing sparse curly hair and sparse eyebrows, relative macrocephaly, hypoplasia of supraorbital ridges, depressed nasal bridge, low set ears, ocular hypertelorism, ptosis and deep philthrum. (c) Face side view showing sparse eyebrows, short nose with depressed nasal bridge and supraorbital hypoplasia.
(a) Bilateral rockerbottomfeet. (b) Bilateral cryptochordism.
Skeletal survey for bone age determination revealed bone age close to and less than 2 years. Routine blood investigations were within normal limits. Serum copper and ceruloplasmin levels, urine for metabolic screening, serum Luteinizing Hormone (LH), Follicle Stimulating Hormone (FSH), testosterone were normal for age. Karyotyping showed genetically male child (46XY). Coagulation profile was normal. Echocardiography revealed dilated right atrium, normal right ventricle and left ventricle, 18mm ostium secundum atrial septal defect with left right shunt, moderate pulmonic stenosis with gradient 35mm of Hg and no features of hypertrophic cardiomyopathy.
Similar characteristic phenotypic features were also found in the elder sibling (aged 9 years) of the baby which over time has become less typical but nonetheless persists. Both the parents were phenotypically normal with no history of similar craniofacial features or developmental delay in their childhood. All of them were found to have normal karyotype.
Diagnosis of Cardiofaciocutaneous Syndrome (CFC) was based on conglomeration of typical sets of clinical features. CFC index, was 14.7 for this baby which fell between 12.1 and 17.3 (mean ± 1 SD) indicating high likelihood of the diagnosis [1]. Molecular genetic testing was beyond our reach. Baby was discharged after 1 week of thorough investigation and with proper counselling regarding the need for multidisciplinary consultation (skin, heart, eye and general paediatrics especially) and regular follow-up. Education was given regarding proper feeding practices, hygiene and increasing social contacts and interaction for improving intellectual development of the baby.
Discussion
CFC syndrome was first termed by Reynolds et al., [2]. Prior to this Blumberg reported 3 individuals with craniofacial dysmorphism and intellectual disability. CFC syndrome is caused due to autosomal dominant de novo mutation of BRAF gene (75-80%) on chromosome 7q34, MAP2K1 & MAP2K2 genes (10-15%) and KRAS gene (<5%) [3]. These genes code for proteins which are part of the RAS/MAPK signalling pathway needed for normal development after birth. Penetrance in CFC syndrome is complete.
CFC syndrome is a rare genetic disorder characterised by distinctive facial features and other abnormalities. Phenotypic features include large forehead with relative macrocephaly, ptosis, depressed nasal bridge, down slanting eyes, ocular hypertelorism, dry, scaly skin with eczema or ichthyosis, sparse curly hair with sparse or absent eyebrows and eyelashes. The babies are born with normal birth weight and length but fail to grow thereafter leading to short stature and failure to thrive except for growth of head (relative macrocephaly). The phenotypic features gradually improves over time, becoming less typical with age [1,4]. There are complaints of feeding difficulties, Gastroesophageal Reflux Disease (GERD), seizures, developmental delay, hypotonia.
The CFC index is generated by attributing scores to these different distinctive phenotypic features and finally summing up the scores. 68% of CFC syndromes have indices between 12.1 and 17.3 (mean± 1SD); 95% will have estimated value between 9.5 and 19.9 (mean ± 2SD) while 99% between 6.9 and 22.5 (mean ± 3SD) [1]. All other related syndromes have scores below 2SD. The index is an important diagnostic aid as well as it helps in differentiating CFC from its two closest differentials, the Noonan and Costello syndromes, distinguished also by their molecular genetics. Noonan is caused due to AD mutation of PTPN11 gene on chromosome 12q24 while Costello syndrome is caused by AD mutation of HRAS gene on chromosome 11p15 [3].
Sparse eyebrows are unlikely in case of Noonan or Costello syndrome. A short nose with broad base is generally absent in Noonan syndrome. Tall forehead with hypertelorism and deep philthrum is unusual in Costello syndrome. Moreover, Noonan syndrome babies have characteristic abnormalities of upper chest (pectuscarinatum) and coagulation disorders leading to easy bruising and bleeding [3]. Costello syndrome babies on the other hand have loose skin on neck, palm and soles with abnormal deep creases. Some Costello syndromes also have papillomata in nares, perioral and anal region with predisposition to childhood tumours [3]. After exclusion of these features, diagnosis of CFC syndrome appeared more likely, based on other noticeable phenotypic features.
More than 100 individuals with CFC syndrome have been reported in the literature. The total number of individuals worldwide with CFC syndrome is estimated to be several hundred [5].
To the best of our knowledge only four cases have been reported from India previously. Nanda et al., reported an 18-year-old girl presenting with dysmorphic facies, dry thickened skin since 6 months of age with dystrophic nails and dysplastic teeth, developmental delay with mild mental retardation and pulmonic stenosis [6]. Thergaonkar et al., described a 18-month-old male child who presented with delayed developmental milestone, characteristic craniofacial abnormality, hyperkeratotic skin, subaortic ventricular septal defect, hepatosplenomegaly with generalised hypotonia [7]. Pavithra et al., reported an one-year-old boy with developmental delay, dysmorphic facies, dry eczematous skin with haemangiomatous plaque, small secundum atrial septal defect with patent ductus arteriosus, mild hepatosplenomegaly and corpus callosum hypoplasia, bifrontal subdural hyperintense areas with prominent frontotemporal subarachnoid space on MRI [8]. Modi et al., described a 30-year-old female with short stature, generalised desquamation of skin with ichthyosis since birth, ostium secundum atrial septal defect and clubbing with digital contractures [9]. In the present case, apart from most of the described features of CFC cases reported from India, additionally cryptochordism, bilateral rockerbottom feet and incomplete grade 2 iris coloboma were present.
Pierpont et al., have given management recommendations for CFC in 2014 [10]. Children and adults with CFC require multidisciplinary care from specialists and the need for comprehensive management has been apparent to families and health care professionals caring for affected individuals. Proper physical and speech therapy as required to be done for promotion of growth, motor and intellectual development.
It was in the year 2002, CFC index was synthesized from available data of 54 patients reported before this time [1]. A good number of further cases have been diagnosed since then. Mutation analysis has also been performed to know the cause in some new cases, but the diagnosis of CFC syndrome is mostly done on clinical basis still now. Sophisticated laboratory and genetic testing is far beyond reach in major part of the globe. Thus, CFC index is still relevant as an aid to diagnosis. We suggest, this index can be updated for diagnosing further cases.
Conclusion
CFC syndrome, though a rare entity, but affected individuals diagnosed worldwide are on the rise. However, mildly affected individuals often remain undiagnosed. These children usually have a good prognosis with a normal life span. We conclude that effort should be made to revise and update the existing CFC index incorporating data from newer cases, so that further cases can be diagnosed with better accuracy.
[1]. Kavamura MI, Peres CA, Alchorne MMA, Brunoni D, CFC index for the diagnosis of cardiofaciocutaneous syndromeAm J Med Genet 2002 112:12-16. [Google Scholar]
[2]. Reynolds JF, Neri G, Herrmann JP, Blumberg B, Coldwell JG, Miles PV, New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement—the CFC syndromeAm J Med Genet 1986 28:413-27. [Google Scholar]
[3]. Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, The cardiofaciocutaneous syndromeJ Med Genet 2006 43:833-42. [Google Scholar]
[4]. Ward KA, Moss C, McKeown C, The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome?Br J Dermatol 1994 131:270-74. [Google Scholar]
[5]. Rauen KA, Cardiofaciocutaneous Syndrome. 2007 Jan 18 [Updated 2016 Mar 3]. In: Pagon RA, Adam MP, Ardinger HH, et al., editorsGeneReviews® [Internet] 1993-2016 Seattle (WA)University of Washington, SeattleAvailable from: http://www.ncbi.nlm.nih.gov/books/NBK1186/ [Google Scholar]
[6]. Nanda S, Rajpal M, Reddy BSN, Cardio-facio-cutaneous syndrome: report of a case with a review of the literatureInt J Dermatol 2004 43:447-50. [Google Scholar]
[7]. Thergaonkar RW, Bhat V, Cardiofaciocutaneous syndromeMedical Journal: Armed Forces India 2013 69(2):175-77. [Google Scholar]
[8]. Pavithra S, Mallya H, Pai GS, Cardiofaciocutaneous syndrome: A rare entityIndian J Dermatol 2012 57:299-301. [Google Scholar]
[9]. Modi R, Patil P, Kothiwale VA, Kamate M, Cardiofaciocutaneous syndromeJ Sci Soc 2014 41:195-96. [Google Scholar]
[10]. Pierpont MEM, Magoulas PL, Adi S, Kavamura MI, Neri G, Noonan K, Cardio-facio-cutaneous syndrome: clinical features, diagnosis and management guidelinesPediatrics 2014 134(4):1149-62. [Google Scholar]