Neurofibromatosis type 1 (NF-1) is an autosomal dominant disorder involving multiple systems and affects approximately 1 out of 3000 persons. Ocular manifestations are lisch nodules, plexiform neurofibroma, optic pathway gliomas. The proper diagnosis of NF-1 is a crucial task for a clinician due to the various clinical manifestations including vision and life threatening malignancies in few patients, which may arise in the different phases of life. The authors report three cases of NF-1, presenting with ophthalmic symptoms in teenager boys. On further ophthalmic and paediatric evaluation the diagnosis of NF-1 was confirmed on the basis of clinical criteria. This series also describe the abnormal facial features like telecanthus and broad nose which has been reported rarely. Case 1 was kept under regular follow-up and Case 2 and Case 3 were planned for the debulking surgery for plexiform neurofibroma of upper eye lid. A multidisciplinary approach is required to diagnose and treat such patients keeping in mind the myriad of clinical manifestations and life-long follow-up is required.
Case 1
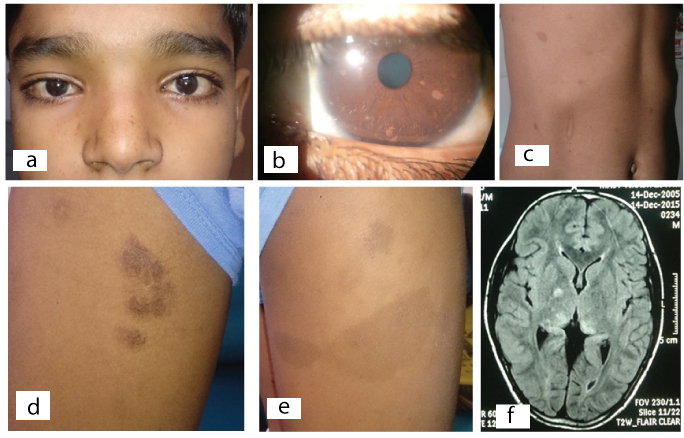
A 13-year-old male child presented to out patient services of Ophthalmology department with the complaints of muddy discolouration and irritation of both eyes. On clinical examination the child was having telecanthus, broad nose with extreme gaze nystagmus [Table/Fig-1a]. His uncorrected visual acuity was 20/20 in both the eyes. Muddy discolouration of conjunctiva and pa-pillary hypertrophy was visible in both eyes. Intra ocular pressure was 14mmHg and 16mmHg in the right and left eye, respectively. On slit lamp examination, cornea was clear and transparent in both eyes, but patient was found to have small multiple hypo-pigmented elevated lesions over the iris suggestive of Lisch nodule in both eyes [Table/Fig-1b]. Pupillary reactions and fundus examination did not reveal any abnormality. Patient was also having multiple pigmented lesions over the trunk [Table/Fig-1c]. Lisch nodule with multiple hyper-pigmented skin lesions prompted us to rule out a neurocutaneous syndrome. Therefore, patient was referred to the paediatric department for further evaluation.
On paediatric evaluation, patient was found to have multiple pigmented flat lesions of 2mm to >5mm in size suggestive of cafe-au-lait spots over the front and back side of the trunk and over the lower extremity [Table/Fig-1c,d]. Two hyper-pigmented elevated lesions, rubbery in consistency one over the left hypochondrium region and another over the inner side of right thigh were present and suggestive of plexiform neurofibroma [Table/Fig-1e]. One neurofibroma was noted over the left external auditory canal. Axillary and inguinal freckling was seen. No neurological deficit was noted. Magnetic Resonance Imaging (MRI) of brain was ordered to rule out any neurological lesion. MRI brain showed the abnormal hyperintense lesion on T2W image over the right side of brain involving basal ganglia region suggestive of Unidentified Bright Objects (UBO) [Table/Fig-1f]. Sphenoid wing dysplasia was not noticed.
Clinical and imaging photographs of a child with NF-1 (Case 1) (a-f). a.) Clinical photograph at presentation showing telecanthus and broad nose; b.) Ocular image showing multiple hypopigmented elevated lesions suggestive of Lisch nodules; c.) Abdominal photograph depicting multiple hyper pigmented macules suggestive of cafe-au-lait spots; d.) Photograph of inner side of thigh showing hairy, elevated rubbery and pigmented lesion suggestive of plexiform neurofibroma; e.) Hyperpigmented lesion over the outer side of thigh suggestive of cafe-au-lai spot; f.) Magnetic resonance imaging (MRI) brain on T2W imaging showing hyper intense lesion in basal ganglia region suggestive of unidentified bright objects.
Based on paediatric, ophthalmological and neurological examination, the diagnosis of Neurofibromatosis type 1 (NF-1) was made. He is the only child of his parents and family history was insignificant for such lesions. Patient was treated for allergic conjunctivitis with topical fluorometholone and carboxy-methylcellulose 0.5%, 4 times per day in both eyes. Patient was kept under observation and asked for an annual examination.
Case 2
A 15-year-old male child presented to Ophthalmology outdoor services with the complaints of huge swelling over the right upper eye lid, brow and forehead area covering the whole right eyeball externally. Patient had history of this swelling since birth which is gradually progressive in nature. The similar swelling behind the left ear pinna was also noted since five years which is gradually increasing in size. No other ocular complaints were noted.
On clinical examination, a localized well defined mass extending from below the medial canthus involving the upper eye lid, bridge of nose, eyebrow area and forehead giving the appearance of S-shaped mechanical ptosis obscuring the right eyeball completely [Table/Fig-2a,b]. The overlying skin over the mass was normal in color. On palpation, the mass was soft in consistency having a bag of worm appearance. The mass behind left ear pinna was also similar in consistency and it was giving the appearance of a dumb-bell shaped lesion [Table/Fig-2c]. Both lesions described above were suggestive of plexiform neurofibroma. Telecanthus and broad nose were also noticed in this patient [Table/Fig-2b]. His best corrected visual acuity was 20/20 in both the eyes. Intraocular pressure was 16mmHg and 18mmHg in the right and left eye, respectively. On slit lamp examination few very small hypopigmented elevated lesions suggestive of Lisch nodules were present [Table/Fig-2d]. Rest of the ocular examination including pupillary reaction and fundus did not show any abnormality. Child was also having multiple pigmented lesions all over the body. Paediatric consultation was sought to rule out any neurocutaneoous syndrome.
Clinical photographs of a child with NF-1 (Case 2) (a-f). a.) Clinical picture showing right eye S-shaped plexiform neurofibroma involving upper eye lid covering the whole eyeball; b.) On retraction of mass telecanthus and normal eyeball is visible; c.) Lateral image showing left plexiform neurofibroma overlying the external ear; d.) Ocular photograph showing fine iris Lisch nodules (arrowheads); e.) Abdominal photograph showing multiple hyperpigmented macules suggestive of cafe-au-lait spots; f.) Picture showing nodule over the lower leg (arrowhead) suggestive of neurofibroma.
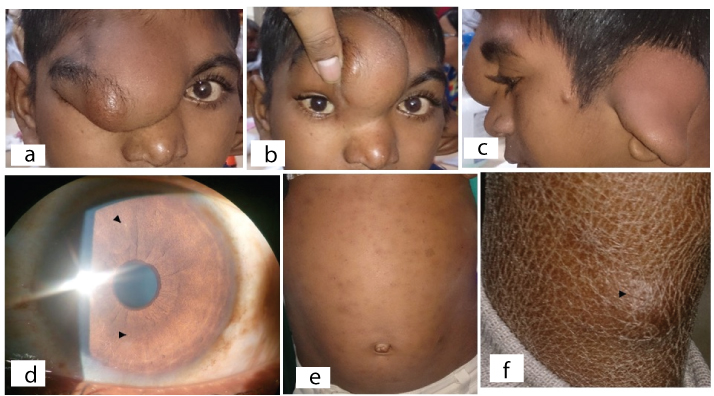
On paediatric evaluation, multiple flat pigmented lesions of 2 mm to >5mm in size suggestive of cafe-au-lait spots were noticed over the trunk [Table/Fig-2e]. Axillary and inguinal region freckling was noticed. No neurological abnormality was seen. One nodular swelling over the back side of lower leg was also seen suggestive of neurofibroma [Table/Fig-2f]. The skin appears dry and scaly over the lower leg due to the frequent rubbing associated with itching around neurofibroma lesion [Table/Fig-2f]. No other systemic abnormality was noticed.
Based on clinical features the diagnosis of NF type I was made. He is the 4th child out of his 7 siblings. His family history was insignificant for such a disease. Patient is currently at the planning phase for MRI and debulking surgery under general anesthesia for upper eye lid plexiform neurofibroma.
Case 3
A 17-year-old male presented with a history of mass over the upper lid since his childhood which was gradually increasing in size and causing drooping of right upper eye lid and blurring of vision. The mass was painless in nature and gradually progressive. Patient also complained of diminution of vision in right eye which worsened in bright daylight. He also developed multiple subcu-taneous nodules and dark pigmented patches all over the body especially at the back of the trunk.
On ophthalmic evaluation a large nodular mass overlying the brow, lid and nose medially which was soft and rubbery in consistency causing mechanical S-shaped ptosis, was observed [Table/Fig-3a]. It was non-tender in nature and was suggestive of plexiform neurofibroma. His uncorrected visual acuity was 20/200 in the right eye and 20/20 in the left eye. Telecanthus and broad nasal bridge were also noticed. Intraocular pressure (Goldmann Applanation Tonometry) was 16mmHg and 18mmHg in the right and left eye, respectively. On slit lamp evaluation multiple hypopigmented iris nodules suggestive of Lisch nodules were visible along with corectopia medially and ectropion uveae [Table/Fig-3b]. Posterior sub-capsular cataract was present in the right eye. Fundus details were not visualized with dense cataract in right eye whereas left eye was normal.
Clinical pictures of a patient with NF-1 (Case 3) (a-e). a.) Clincal photograph showing right upper eye lid boggy swelling suggestive of plexiform neurofibroma causing S-shaped mechanical ptosis (black arrowhead); b.) Slit lamp photograph showing multiple iris Lisch nodules (blue arrow) and corectopia; c.) Multiple small subcutaneous nodules; d.) Subcutaneous nodule (upper arrow) and multiple cafe-au-lait spots (lower arrow) over the back; e.) A large cafe-au-lait (blue arrow) spot over the left lateral side of trunk with multiple subcutaneous nodules.
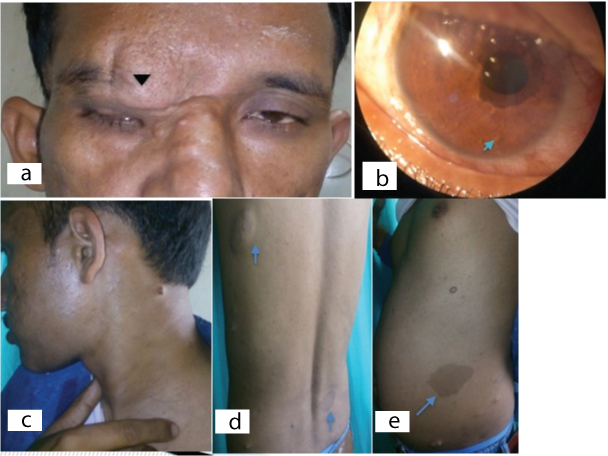
On systemic examination patient was having axillary freckling, multiple nodular lesions suggestive of neurofibroma [Table/Fig-3c], and multiple hyperpigmented macules (cafe-au-lait spots) with sizes varying from 2mm to >5mm over the trunks and limbs [Table/Fig-3d]. Several soft cutaneous sessile neurofibromas varying from few mm to several cm in diameters were present along the trunk, limbs and neck region [Table/Fig-3e]. MRI brain was ordered to rule out optic glioma, meningioma, acoustic neuroma, schwanoma and it did not show any abnormal feature. Sphenoid wing dysplasia was also not present.
On the basis of the above mentioned features the diagnosis of NF-1 was made and patient was planned for debulking surgery for the large plexiform neurofibroma of the upper eye lid.
Consent: Consent was obtained from all the patients for publication.
Discussion
Neurofibromatosis (NF) is the commonest neurocutaneous syndrome which involves skin and central nervous system. It is inherited in an autosomal dominant fashion affecting 1 out of 3,000 persons caused due to mutations of NF-1 gene in chromosome region 17q11.2, encoding neurofibromin protein [1,2]. It is characterized by the presence of cafe-au-lait spots, axillary or inguinal freckiling, iris Lisch nodules, neurofibromas, optic nerve glioma and sphenoid dysplasia [3]. Abnormal facial features in NF-1 has been reported rarely [4]. We described the rare cases of NF-1 with abnormal facial features which were presented with ophthalmic symptoms.
NF-1 diagnosis is based on the presence of atleast 2 of the 7 criteria which include: six or more cafe-au-lait macules over 5mm in greatest diameter in prepubertal individuals, frecklings over the axillary and inguinal area, Lisch nodules over the iris, two or more neurofibroma or one plexiform neurofibroma, [sphenoid dysplasia], optic glioma and first-degree relative with NF-1 [5]. All of our cases met with 3 or more criteria Case 1: Cafe-au-lait macules, axillary or inguinal freckling, Lisch nodules; Case 2: Cafe-au-lait macules, axillary or inguinal freckiling, Lisch nodules, plexiform neurofibroma; Case 3: Plexiform neurofibroma, Lisch nodules, axillary freckling, cafe-au-lait macules).
The ophthalmological features with NF-1 were studied in 72 consecutive patients with NF-1 in a study and it was found that the ophthalmological features are relatively rare [6]. NF-1 presenting with ophthalmic associations like eyelid plexiform neurofibroma, optic glioma, primary congenital glaucoma have been described in the literature [7–9]. Choroidal nodules, vasoproliferative tumours, unusual limbal lesion and neovascular glaucoma are the other ophthalmic manifestations that have been reported rarely [10–13]. Case 1 presented with non-specific ocular complaints which was diagnosed as a case of allergic conjunctivitis. On further ophthalmic evaluations Lisch nodules were seen in both eyes. We reported the abnormal facial features like telecanthus, broadening of nose which has been reported rarely [4]. A distinctive facial appearance in the children with NF-1 was first reported by Kaplan et al., in their case series [4]. These children were having telecanthus, antimongoloid slant of the palpebral fissure, broad nose and tapering chin [4]. The morphological features were similar to Noonan syndrome but the manifestations in children were sufficiently different to label it as a variant of Noonan syndrome. Similarly, the poor evidence for the association of Noonan phenotype and NF-1 was found in a series of 453 cases with NF-1 [14]. However, in a series of seven patients with variable phenotype of NF-1 and Noonan syndrome, the mutation was found on NF-1 gene which represents it as a variant of NF-1 cases [15]. Therefore, it is important to keep in mind the abnormal facial features during the work-up of a patient with NF-1. Plexiform neurofibroma of upper eye lid and external ear was seen in Case 2. A similar dumb-bell shaped neurofibroma over the external ear was reported by Shirol et al., [16]. Nystagmus has been reported as the initial presenting sign of chiasmal glioma in young children in a series of 22 patients by Toledano et al., [17]. Extreme gaze nystagmus was noticed in Case 1 which was not associated with any optic pathway glioma.
Debulking of tumour mass is needed in cases of plexiform neurofibroma with NF-1. Ptosis surgery and lateral canthal fixation are the other treatment modalities in cases of plexiform neurofibroma. The clearance of visual axis and cosmetic appearance are the basic goals for surgery. An acceptable result can be observed with lid and oculoplastic surgeries as shown in earlier studies [18,19]. Glaucoma surgery is required in cases of NF-1 associated with glaucoma [20].
This series represents the various clinical manifestations of NF-1 and highlights the importance of ophthalmic examination in such cases as they first presented to the ophthalmologist. As the disease requires a life long follow-up for the various clinical manifestations which may arise in the different phase of life, the early diagnosis of NF-1 becomes a crucial task for clinicians. A holistic multidisciplinary approach is required to diagnose and treat these patients. This case series provide the further knowledge about clinical features of patients with NF-1 which might be helpful for further understanding about the disease.
Conclusion
Since the patients with NF-1 can present with ophthalmic complaints, the ophthalmic evaluation is an essential part of examination. Proper diagnosis and treatment require the understanding about the myriad of manifestations of NF-1.
[1]. Huson SM, Compston DA, Clark P, Harper PS, A genetic study of von Recklinghausen neurofibromatosis in south east Wales IJ Med Genet 1989 26:704-11. [Google Scholar]
[2]. Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locusCell 1990 62:187-92. [Google Scholar]
[3]. McGaughran J, Harris DI, Donnai D, Teare D, MacLeod R, Wester Beek R, A clinical study of type 1 neurofibromatosis in North West EnglandJ Med Genet 1999 36:197-203. [Google Scholar]
[4]. Kaplan P, Rosenblatt B, A distinctive facial appearance in neurofibromatosis von Reckling-hausenAm J Med Genet 1985 21:463-70. [Google Scholar]
[5]. Stumpf D, Alksne J, Annegers J, Neurofibromatosis. Conference statement. National institute of health consensus development conferenceArch Neurol 1988 45:575-78. [Google Scholar]
[6]. Ruggieri M, Pavone P, Polizzi A, Di Pietro M, Scuderi A, Gabriele A, Lannetti P. Ophthalmological manifestations in segmental neurofibromatosis type 1Br J Ophthalmol 2004 88:1429-30. [Google Scholar]
[7]. Tokraz-Sawinska E, Puchalska-Niedbal Lachowicz E, Litwinska J, Plexiform neurofibroma of the upper eyelid and orbit in a patient with von Recklingausen disease—case reportKlein Oczena 2014 116:267-71. [Google Scholar]
[8]. Levin MH, Armstrong GT, Broad GH, Zimmerman R, Bilaniuk LT, Feygin T, Risk of optic pathway glioma in children with neurofibromatosis type 1 and optic nerve tortuosity or nerve sheath thickeningBr J Ophthalmol 2016 100(4):510-14. [Google Scholar]
[9]. Li H, Liu T, Chen X, Xie L, A rare case of primary congenital glaucoma in combination with neurofibromatosis 1: A case reportBMC Ophthalmol 2015 15:149 [Google Scholar]
[10]. Abdolrahimzadeh S, Felli L, Plateroti R, Plateroti AM, Giustini S, Calvieri S, Morphological and vasculature features of the choroid and associated choroid-retinal thickness alteration in neurofibromatosis type-1Br J Ophthalmol 2015 99:789-93. [Google Scholar]
[11]. Shields JA, Pellergrini M, Kaliki S, Mashayekhi A, Shields CL, Retinal vasoproliferative tumours in 6 patients with neurofibromatosis type 1JAMA Ophthalmol 2014 132:190-96. [Google Scholar]
[12]. Samarawickrama C, Cherepanoff S, Di Girolamo N, Benger R, Watson S, Unusual limbal lesion in neurofibromatosis type 1Cornea 2015 34:347-49. [Google Scholar]
[13]. Pichi F, Morara M, Lembo A, Ciardella AP, Meduri A, Nucci P, Neovascular glaucoma in-duced by peripheral retinal ischemia in neurofibromatosis type 1: management and imaging featuresCase Rep Ophthalmol 2013 4:69-73. [Google Scholar]
[14]. Colley A, Donnai D, Evans DG, Neurofibromatosis/Noonan phenotype: a variable feature of type 1 neurofibromatosisClin Genet 1996 49:59-64. [Google Scholar]
[15]. Huffmeier U, Zenker M, Hoyer J, Fahsold R, Rauch A, A variable combination of features of Noonan syndrome and neurofibromatosis type 1 are caused by mutations in the NF1 geneAm J Med Genet A 2006 140:2749-56. [Google Scholar]
[16]. Shirol SS, Kodaganur S, Dani AA, Bhagwat PV, Dumbbell-shaped neurofibroma over the external earJ Cutan Aesthet Surg 2015 8:175-77. [Google Scholar]
[17]. Toledano H, Muhsinoglu O, Luckman J, Goldenberg-Cohen N, Michowiz S, Acquired nys tagmus as the initial presenting sign of chiasmal glioma in young childrenEur J Paediatr Neurol 2015 19:694-700. [Google Scholar]
[18]. Altan-Yayicioglu R, Hintschich C, Clinical features and surgical management of orbitotem-poral neurofibromatosis: A retrospective interventional case seriesOrbit 2010 29:232-38. [Google Scholar]
[19]. Lee V, Ragge NK, Collin JR, Orbitotemporal neurofibromatosis. clinical features and surgical managementOphthalmology 2004 111:382-88. [Google Scholar]
[20]. Morales J, Chaudhry IA, Bosley TM, Glaucoma and globe enlargement associated with neurofibromatosis type 1Ophthalmology 2009 116:1725-30. [Google Scholar]