Introduction
More than 40 out of 50 best-selling pharmaceutical products in the US and European markets are given orally [1]. S-1 (Taiho Pharmaceutical, Tokyo, Japan) is a novel oral fluoropyrimidine derivative with single-agent response rates more than 40% for Advanced Gastric Cancer (AGC) [2]. This new oral anti-tumour drug contains a combination of 3 pharmacological agents (at a molar ratio of 1:0.4:1)-Tegafur (FT), which is a prodrug of 5-Fluorouracil (5-FU); 5-Chloro-2,4-Dihydroxypyridine (CDHP), which inhibits the activity of Dihydropyrimidine Dehydrogenase (DPD) activity; and Potassium Oxonate (Oxo), which reduces the Gastrointestinal (GI) toxicity of 5-FU. The [Table/Fig-1] shows the mode of action of S-1 with their major adverse reactions [3].
Mode of action of S-1 [3].
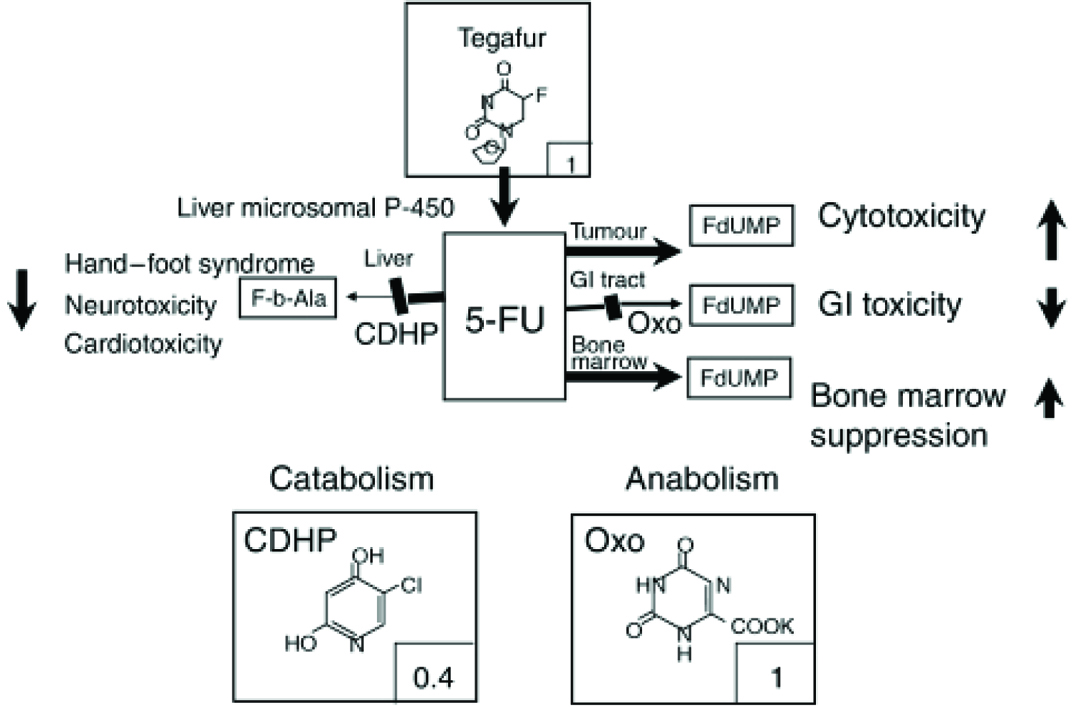
It is widely used for treating gastric, pancreatic, lung, head, neck, and breast carcinomas. FT is a prodrug that is converted by liver Cytochrome P4502A6 (CYP2A6) to 5-FU. 5-FU is then actively catabolized by liver into inactive metabolites. About 90% of FU is converted to inactive metabolites by enzyme DPD. Hence, remaining 10% would be responsible for anti-tumour activity. It owes its anti-tumour properties to the production of 5-FU, which is gradually converted from FT, following oral administration of S-1. Both CDHP and potassium Oxo act as bio-modulators to impart better anti-cancer effect of 5-FU and do not have any anti-tumour activity by themselves. CDHP inhibits DPD enzyme, that is responsible for inactivation of 5-FU and is 180 times more active than uracil in-vitro. Potassium Oxo, which is Orotate Phosphoribosyl-Transferase (OPRT) inhibitor, is found in higher concentration in GI tract, which helps in decreased formation of 5-FU nucleotides (metabolites), thereby, decreasing the GI toxicity as diarrhea and stomatitis [4].
The additional value of expression of the OPRT gene over DPD expression in prediction of the response of chemotherapy was clearly demonstrated in gastric cancer patients treated with S-1 [5]. The low level of DPD, Thymidylate Synthase (TS) activities and a high level of OPRT activity enhance the anti-tumour effects of 5-FU and S-1. Gastric cancer is the second most frequent cause of cancer-related mortality worldwide, accounting for approximately 700,000 deaths annually [6]. The idea of biochemical modulation was adopted to the treatment of gastric cancer as the third generation of chemotherapy. Combinations of 5-FU and high- or low-dose cis-platinum (CDDP), high-dose fluorouracil (UFT), S-1, and capecitabine are included in this category [7].
Oxo is distributed in GI tract at a high concentration following oral administration and it prevents phosphorylation (i.e., activation) of 5-FU by inhibiting the effect of OPRT [8]. 5-FU was originally synthesized in Research and Development Division of National Cancer Institute. After the introduction of 5-FU, 5-FU and its analogues were found to have inhibitory effect in the mammalian cells, micro-organisms and plant cells. This agent was developed as the first generation chemotherapeutic agent, active for gastric cancer. FAM (5-Fu, Doxorubicin, Mitomycin–C) was the first generation chemotherapy, commonly used in the 1980s [9]. FAMTX (5-Fu, Doxorubicin and Methotrexate), EAP {Etoposide, Adriamycin (Doxorubicin), Cisplatin}, PELF (Cisplatin, Epirubicin, Leucovorin preceding Fluorouracil) were the second generation chemotherapy used in the late 1980’s. Second generation therapy was more effective than the first generation combination therapy, but still the response rate was not sufficient and the Adverse Effects (AEs) observed could not be overlooked [10]. Hence, there was a need to develop a more selective and efficient therapy. High dose infusional 5-FU/Leucovorin (HDFL) combination chemotherapy eventually proved to be the third generation therapy.
The search for a still better chemotherapy continued as a result of which the idea of biochemical modulation came and chemotherapeutic regimens with more effectiveness, tumour selective properties and high response rates with decreased AEs were developed. Combination of 5-FU with cisplatin (CDDP), UFT and capecitabine and the most recent oral S-1 combinations was representative of this category.
Clinical Studies
Comparing the anti-cancer effects of three agents S-1 showed better anti-tumour effect with less adverse effects like stomatitis, depilation, body weight loss and diarrhea [11]. S-1 was significantly effective in inhibiting tumour growth in nude mice implanted with human stomach, breast, head, neck, pancreatic and lung cancer. Mori et al., also demonstrated that S-1 showed more therapeutic effect in prevention of gastric cancer [12]. Based on this, phase I and phase II studies of S-1 were carried out. Initial phase I study was started in Japan (in 1992), Europe and Phase II trials were conducted in the US. In Japan, S-1 was shown to have a great response in the treatment of several cancers and it also offered the Quality of Life (QoL), ease of administration and safety characteristics as it lowered the AEs and had lower incidence of grade 3-4 toxicity. Based on good results observed in patients with gastric cancer, Ministry of Health and Welfare of Japan granted permission for the manufacturing of S-1 in January 1999 [12].
The phase I-II studies on S-1 and combination therapy fairly demonstrated that S-1 is non-inferior to 5-FU continuous infusion whereas response rate and progression free survival were found to be better than 5-FU. Two late phase II studies of S-1 as the single agent in AGC in Japan showed the response rate of 44% and 49% respectively [13].
Depending on the promising results shown by phase I and II studies same study was carried up to phase III level. JCOG9912 trial proved that S-1 was non-inferior to the continuous infusion of 5-FU in terms of Overall Survival (OS). The combination therapy with S-1 was in progress to obtain a better survival benefit, lower cost and better QoL [14]. [Table/Fig-2] represents the comparative results of S-1 [12,14–17]. Two randomized phase III trials were reported from Japan till date [17,18].
Comparative results seen with S-1 experiments.
| References | Level of evidence | Findings |
|---|
| Mori et al., 2006 [12] | III | S1 showed more therapeutic effect in prevention of gastric cancer. |
| Boku et al., 2009 [17] | I | S-1 was non-inferior to the continuous infusion of 5-FU in terms of Overall Survival (OS). |
| Shiozawa et al., 2009 [14] | I | Japanese randomized phase III trial (GC0301/TOP-002) conducted in 326 patients to compare S1 with S-1 plus irinotecan showed a significant response advantage for combination arm (41.5% vs. 26.9%, p=0.35). |
| Zang et al., 2009 [15] | I | In multicenter phase II clinical trial, the efficacy of oral S1 with biweekly regimen paclitaxel showed that response rate was good (43.6%) with greater survival days with this treatment regimen. |
| Baba et al., 2009 [16] | I | A phase I trial applying increasing doses of oral administration of S-1 (65-80 mg/mL) for 21 days and increasing doses of CDDP (60-80 mg/mL) on day 22, every 35 days, was conducted to determine MTD and the recommended dose (RD) for the phase II studies. Phase I study of the sequential administration of S-1 and cisplatin for metastatic gastric cancer showed that this sequential administration of S-1 and CDDP every 35 days was tolerable and warranted a phase II trial. |
Combination Regimens with S-1
Combination with Cisplatin (CDDP)
Cisplatin is a platinum base chemotherapeutic agent, which causes apoptosis by cross-linking of DNA. Phase I pharmacokinetic study of S-1 and Cisplatin in patients with AGC showed that Mean Tolerated Dose (MTD) of S-1 (25mg/mL administered twice daily from day 1 to 21) and Cisplatin (75mg/mL administered in day 1) combination varied in the Western and Asian patients due to the difference in the metabolism of CYP2A6. Hence, MTD for the Western patients was lower than in Asian patients since in the Western patients there was more activity of CYP2A6 converting 5-FU at a higher ratio achieving higher AUC than Asians [19].
S-1 as single therapy had the response rate of 26-45%, while S-1 combined with Cisplatin showed 76% response rate in the Japanese study of 25 patients. The combination of S-1 (80mg/mL for 21 consecutive days and Cisplatin 50mg/mL on day 8) was effective neo-adjuvant therapy in highly AGC patients. The response rate was more than 60% and grade 3-4 AEs were less than 10% [20]. A phase I trial applying increasing doses of oral administration of S-1 (65-80mg/mL) for 21 days and increasing doses of CDDP (60-80-mg/mL) on day 22, every 35 days, was conducted to determine MTD and the Recommended Dose (RD) for the phase II studies. Phase I study of the sequential administration of S-1 and cisplatin for metastatic gastric cancer showed that this sequential administration of S-1 and CDDP every 35 days was tolerable and warranted a phase II trial. The RD was a combination of S-1 (80mg/mL) and CDDP (70mg/mL) [16]. S-1 and Cisplatin were shown to be more efficient than other combinations in the further phase II and phase III studies. Phase II studies were performed by Taiho pharma using S-1 in combination with Cisplatin as the first line therapy in advanced non-small cell lung cancer [21].
After the encouraging results of combination therapy of S-1 and Cisplatin, phase III trial was conducted with two arms- Arm A: S-1 (80mg/mL for 28 days, followed by 14 days rest), and Arm B: S-1 (80mg/mL for 21 days, followed by 14 days rest and Cisplatin iv on day 8). Altogether, 305 patients, with 152 and 153 in arm A and B respectively were randomized for the study. OS rate was superior in Arm B compared to Arm A. Response rate in Arm A was 31.1% and 54% in Arm B [22,23]. The AEs observed were neutropenia, leucopenia, anaemia, nausea, anorexia, and no treatment related death. Hence, it was concluded that the combination regimen was effective and well tolerated and could be further studied to establish it as the first line standard therapy for AGC. The other study called SPIRITS trial suggested that S-1 plus CDDP was superior to S-1 alone with response rates of 54% to 31% and OS of 13 months to 11 months [22,23].
For combined treatment with docetaxel, cisplatin and S-1 in patients with AGC, recommended dose was docetaxel (40mg/mL), cisplatin (70-mg/mL) and S-1 (80mg/mL/day). This regimen yielded a high rate of tumour response and could be administered safely. Phase II studies of this regimen are under investigation. Besides gastric cancer, Phase II trial of the concurrent chemo-radiotherapy with S-1 plus cisplatin were also conducted in patients with unresectable locally advanced squamous cell carcinoma of head and neck, and advanced non-small cell lung cancer [24,25].
Combination with Irinotecan (CPT-11)
In phase I and phase II studies carried out in Japan S-1 was administered from 1-14 days at 80mg/mL and CPT-11 was administered as 80mg/mL, and increased up to 100mg/mL on days 1 and 8, and repeated in each three weeks. The RD of CPT-11 was 80mg/mL. After phase II study in patients with no prior chemotherapy, it was observed that the combination therapy had no considerable increase in response rate but had a greater tumour control rate and also slightly increased the survival rate. The adverse effect of grade 3 or worse was low. Hence, the combination therapy is now under greater consideration for the advanced and recurrent cancers [26].
Japanese randomized phase III trial (GC0301/TOP-002) was conducted in 326 patients to compare S-1 with S-1 plus irinotecan. S-1 monotherapy included S-1 80mg/mL/day throughout days 1-28, repeated every 6 weeks. The S-1 and irinotecan regimen included S-1 80mg/mL from 1-21 days and irinotecan 80mg/mL on days 1 and 15, followed by a 2 weeks rest. The result showed a significant response advantage for combination arm (41.5% vs. 26.9%, p=0.35). A greater toxicity without neutropenia, anorexia and diarrhea was observed without any significant OS difference [21]. This might probably be an indication that further modulations and deeper studies in this combination therapy could have been mandatory. Proper management of the AEs and clinically more beneficial combination would bring a revolution. Irinotecan and S-1 combination did not show any superiority. Hence, a more suitable combination therapy could be attached with other anti-tumour agent. A phase III trial was carried out with irinotecan plus 5-FU/Leucovorin and 5-FU and Cisplatin in 337 patients with gastric cancer. The result indicated no significant differences in terms of OS, QoL and toxicity [14].
The predictive values of TS and DPD gene expressions were retrospectively evaluated in patients with AGC treated by a regimen containing S-1. Twenty six patients were treated with S-1 combined with irinotecan (CPT-11). With the focus on molecular level, real time reverse transcription PCR is the suitable tool to measure gene expression of TS and DPD in primary tumours. Results showed that treatment effects of S-1 monotherapy for AGC were determined by the status of TS gene expression, regardless of DPD gene expression. TS predictive power was overcome by CPT-11 combination therapy with S-1 [27].
Combination with Taxanes
A successful treatment of AGC was done by department of digestive surgery and surgical oncology in Japan by using the combination of S-1 and paclitaxel as neo-adjuvant chemotherapy. A 67-year-old patient with AGC, ascites and large lymph node metastasis, adjacent to pancreas, was treated with oral S-1 80mg/mL, administered twice daily after meals for 2 weeks and paclitaxel 100mg/ml i.v. on day 1 and 15 only. After receiving 2 cycles of this regimen separated by a 14-day interval, the positive response was clearly observed by endoscopy and CT scanning. Endoscopy showed the considerable decrease in primary lesion and shrinkage of gastric wall. CT scanning showed that there were no signs of ascites and large metastatic lymph nodes after chemotherapy. The combination therapy in this case assisted in decreasing the tumour load prior to resection. No severe AEs was observed, patient experienced grade 2 leucopenia and grade 1 nausea [28]. The future aspect of the study might include the complete treatment of advanced tumours by combined chemotherapy. In a phase II study, the activity and safety of biweekly paclitaxel and oral S-1 as treatment for unresectable and recurrent gastric cancer was assessed. Chemotherapy was performed using two anticancer agents, S-1 and paclitaxel. Oral S-1 (80 mg/mL) was administered twice a day after meals for two consecutive weeks from day 1 to 14, followed by a 2 week recovery period; paclitaxel (120 mg/mL) was administered i.v., biweekly, on days 1 and 15. The patient received cycles of this regimen every 4 weeks (28-day cycles). The primary end point was the response rate according to the response evaluation criteria in solid tumours. The most common treatment-related Grade 3/4 AEs were neutropenia (37.5%), appetite loss, diarrhea, decreased sodium (each 5%) and anaemia, increased Alanine Aminotransferase (ALT), generalized fatigue and dizziness (each 2.5%). Almost all the patients experienced alopecia. In multicenter phase II clinical trial, the efficacy of oral S-1 with biweekly regimen paclitaxel is assessed for unresectable and recurrent gastric cancer. The result showed that response rate was good (43.6%) with greater survival days with this treatment regimen [15].
In the study, to determine the Dose-Limiting Toxicity (DLT) and activity of combination with docetaxel and S-1 on unresectable gastric cancer, docetaxel was administered i.v. on day 1 and S-1 was administered orally on days 1-14, every 3 weeks. Doses of each drug in phase I study were docetaxel 60-75mg/mL and S-1 60-80mg/mL. Combination with docetaxel and S-1 was active against AGC and presented manageable toxicities [29].
In another phase III study, the final results showed that S-1 plus docetaxel had good clinical activity with acceptable toxicity in patients with advanced or recurrent gastric cancer. Major toxicities were leucopenia (52.3%), alopecia (46.5%), neutropenia (45.3%) and anorexia (41.8%). Grade 3/4 haematologic toxicities included neutropenia (36.0%), leucopenia (31.7%), febrile neutropenia (4.7%) and anaemia (1.2%), which occurred in 55.6% (40/72) within three cycles [30].
The study proved that the combination regimen was acceptable as the first line therapy in advanced or recurrent gastric cancer. So, this combination regimen might prove to be the preferred first line chemotherapy.
Pharmacokinetic Perspective
The plasma Cmax values of FT, 5-FU, Oxo acid and CDHP increased in dose-dependent manner. Pharmacokinetics of CDHP and uracil were linear over the dose range. Cumulative urinary excretion of FT was predominantly as 5-FU [31].
Conversion to 5-FU may occur predominantly in tumour cells and liver by CYP2A6, although a role for thymidine phosphorylase was also postulated [32]. DPD catalyzes the degradation of 5-FU and is responsible for more than 80% of its elimination. Combining CDHP with 5-FU serves several purposes. First, CDHP is a potent reversible competitive inhibitor of DPD, preventing degradation of 5-FU but it has no direct effect on 5-FU anabolism. Second, in combination with oral FT, CDHP enhances plasma 5-FU derived from FT. Co-administration of CDHP with FT also increases the 5-FU concentrations in the tumour, 5 to 10 times compared to FT alone or 5-FU itself [29].
Oxo accumulates specifically in normal gut in which it inhibits the conversion of 5-FU to 5-fluorouridine 5’-monophosphate, a precursor of 5-fluoro-2’-deoxyuridine-5’-monophosphate (5-FDUMP), the active metabolite of 5-FU, which inhibits the target enzyme TS. Since Oxo does not accumulate in the tumour, it has a selective protective effect against GI toxicity, but does not affect anti-tumour activity. Oxo does not affect the pharmacokinetics of 5-FU [33].
Briefly, the DLT consists of diarrhea, with grade 3–4 diarrhea in 3 of 5 patients treated at the highest dose level and in 4 of 11 patients treated at 40mg/mL. However, toxicity is predominantly observed in heavily pre-treated patients. At 45-mg/mL, only one course could be completed because toxicity was too severe to continue treatment in subsequent courses as oral administration of S-1 resulted in a high bio-availability of FT and CDHP and that FT was rapidly converted to the active drug 5-FU, but initially not to its catabolite F-b-Ala. The dose of S-1 was linearly correlated with the Cmax and AUC of FT, CDHP and Uracil. After a single administration of S-1, there was no correlation between the Cmax or AUC of oxonic acid and the dose of S-1, but there was a linear correlation between oxonic acid at 2 hour and the dose of S-1 during the first course and during subsequent courses. In addition, there was an up to fourfold accumulation of plasma oxonic acid during treatment [34].
In European patients some plasma accumulation of oxonic acid occurred during the twice-daily administration of S-1, but accumulation of oxonic acid in the gut was not sufficient to provide adequate protection against damage to the gut mucosa, which was considered to be responsible for diarrhea. The uptake and metabolism of FT are determined by a number of different parameters. First, after oral intake, FT has to be absorbed from GI tract, after which it is rapidly distributed. Subsequently, it has to be cleaved to 5-FU, for which CYP2A6 is responsible [35]. This enzyme is widely distributed in Caucasians but has a lower expression in Japanese [36]. The subsequent fate of 5-FU is determined by its distribution and elimination. The latter process is influenced by the extent of inhibition of 5-FU degradation by DPD. The more potent this inhibition, the longer will be the retention of 5-FU in plasma. So, theoretically, one would expect a relatively short half-life of 5-FU derived from FT in the UFT combination compared to that in the S-1 combination. This has indeed been found, since, 5-FU derived from UFT has a shorter half-life of 40 minutes compared to 2–4 hours for FU derived from S-1 [32]. Ethinyl uracil is a more potent inhibitor of DPD leading to an even longer retention of 5-FU, despite the fact that in this formulation, 5-FU itself was given and not FT [36]. The initial half-life of 5-FU derived from FT given as an Intra Venous (IV) infusion is 10 minutes and is comparable to that of 5-FU given as an Intra Venous (IV) infusion but is significantly shorter than that with each DPD inhibitor, including the relatively weak DPD inhibitor uracil [32]. Thus, it can be concluded that the increased half-life of 5-FU derived from UFT and S-1 is due to the inhibition of DPD and that the difference between the S-1 formulation and ethinyl uracil/5-FU seems to be related to the better efficacy of the suicide DPD inhibitor, ethinyl uracil. The latter aspect, the reversible inhibition by CDHP and the suicide inhibition by FU, can be evaluated by the difference in retention of DPD inhibition. DPD levels in WBC of treated patients even increased during treatment, although this did not seem to affect the efficacy of DPD inhibition, based on accumulation of uracil. This reversibility of CDHP can also be judged from the appearance of F-b-Ala in the urine, which increased when the concentration of CDHP decreased. This gives CDHP several advantages compared to uracil in UFT and ethinyl uracil in the ethinyl uracil/5-FU combination. The inhibition by CDHP is quite potent but easily reversible upon withdrawal. Therefore, no severe toxicity is expected when patients, who discontinue S-1 treatment, receive another 5-FU-based chemotherapy [36].
Conclusion
Though, the introduction of third generation chemotherapy showed high response rates, DLTs and side effects were still not considerable and frequent infusion led to patients’ non-compliance. The search for a still better chemotherapy continued. S-1 is a novel oral fluoropyrimidine derivative, widely used for treating gastric, pancreatic, lung, head, neck, and breast carcinomas. The combination therapy with S-1 has been in progress to obtain a better survival benefit, lower cost and better QoL.
Sources of Funding: This research did not receive any grant from any public or commercial funding agency, or not-for-profit sectors.
[1]. Lennernas H, Abrahamsson B, The use of biopharmaceutic classification of drugs in drug discovery and development:current status and future extensionJ Pharm Pharmacol 2005 57(3):273-85. [Google Scholar]
[2]. Koizumi W, Nakano S, Hasegawa K, Phase II studyof S-1, a novel oral derivative of 5-fluorouracil, in advanced gastric cancer: for the S-1 Cooperative gastric cancer study groupOncology 2000 58:191-97. [Google Scholar]
[3]. Saif MW, Syrigos KN, Katirtzoglou NA, S-1: A promising new oral fl -uoropyrimidine derivativeExpert OpinInvestig Drugs 2009 18(3):335-48. [Google Scholar]
[4]. Chuah B, Goh B, Lee S, Soong R, Lau F, Mulay M, Comparison of the pharmacokinetics and pharmacodynamics of S-1 between Caucasian and East Asian patientsCancer Sci 2011 102:478-83. [Google Scholar]
[5]. Ichikawa W, Suto K, Simple combina-tions of 5-FU pathway genes predict the outcome of metastaticgastric cancer patients treated by S-1Int J Cancer 2006 119:1927-33. [Google Scholar]
[6]. Parkin DM, Ferlay J, Global cancer statistics in the year CACancer J Clin 2002 55:74-108. [Google Scholar]
[7]. Koizumi W, Ujiie S, A pilot phase II study of capecitabine in advanced or recurrent gastric cancerOncology 2003 64:232-36. [Google Scholar]
[8]. Saif MW, Syrigos KN, Katirtzoglou NA, S-1: a promising new oral fluoropyrimidine derivativeExpert OpinInvestig Drugs 2009 18(3):335-48. [Google Scholar]
[9]. Shirasaka T, Development history and concept of an oral anticancer agent S-1(TS-1): its clinical usefulness and future vistasJpn J Clin Oncol 2009 39(1):2-15. [Google Scholar]
[10]. Kobayakawa M, Kojima Y, Tegafur/gimeracil/oteracil (S-1) approved for the treatment of advanced gastric cancer in adults when given in combination with cisplatin: a review comparing it with other fluoropyrimidine-based therapiesOncoTargets and Therapy 2011 4:193-201. [Google Scholar]
[11]. Shirasaka T, Nakano K, Takechi T, Satake H, Uchida J, Fujioka A, Antitumour activity of 1 M tegafur-0.4 M 5-chloro-2,4-dihydroxypyridine-1 M potassium oxonate (S-1) against human colon carcinoma orthotopically implanted into nude ratsCancer Res 1996 56(11):2602-06. [Google Scholar]
[12]. Mori S, Kishimoto H, Tauchi K, Higuchi K, Histological complete response in advanced gastric cancer after 2 weeks of S-1 administration as neoadjuvant chemotherapyGastric Cancer 2006 9(2):136-39. [Google Scholar]
[13]. Koizumi NJ, Inatomi TJ, Sotozono CJ, Fullwood NJ, Quantock AJ, Kinoshita S, Growth factor mRNA and protein in preserved human amniotic membraneCurr Eye Res 2000 20(3):173-77. [Google Scholar]
[14]. Shiozawa M, Sugano N, Tsuchida K, Morinaga S, Akaike M, Sugimasa Y, A phase I study of combination therapy with S-1 and irinotecan (CPT-11) in patients with advanced colorectal cancerJ Cancer Res ClinOncol 2009 135(3):365-70. [Google Scholar]
[15]. Zang DY, Yang DH, Lee HW, Hwang SW, Song HH, Jung JY, Phase I/II trial with docetaxel and S-1 for patients with advanced or recurrent gastric cancer with consideration to ageCancer ChemotherPharmacol 2009 63(3):509-16. [Google Scholar]
[16]. Baba E, Fujishima H, Kusaba H, Esaki T, Ariyama H, Kato K, Tanaka R, Mitsugi K, Shibata Y, Harada M, Nakano S, Phase I study of the sequential administration of S-1 and cisplatin for metastatic gastric cancerAnticancer Res 2009 29(5):1727-32. [Google Scholar]
[17]. Boku N, Yamamoto S, Fukuda H, Shirao K, Doi T, Sawaki A, Gastrointestinal oncology study group of the japan clinical oncology group: Fluorouracil versus combination of irinotecan plus cisplatin versus S-1 in metastatic gastric cancer: a randomised phase 3 studyLancet Oncology 2009 10:1063-69. [Google Scholar]
[18]. Takashima A, Boku N, Kato K, Nakamura K, Mizusawa J, Fukuda H, Survival prolongation after treatment failure of first-line chemotherapy in patients with advanced gastric cancer: combined analysis of the Japan clinical oncology group trials JCOG9205 and JCOG9912Gastric Cancer 2014 17(3):522-28. [Google Scholar]
[19]. Ajani JA, Faust J, Ikeda K, Yao JC, Anbe H, Carr KL, Phase I pharmacokinetic study of S-1 plus cisplatin in patients with advanced gastric carcinomaJ Clin Oncol 2005 23(28):6957-65. [Google Scholar]
[20]. Yoshikawa T, Omura K, Kobayashi O, Nashimoto A, Takabayashi A, Yamada T, A phase II study of preoperative chemotherapy with S-1 plus cisplatin followed by D2/D3 gastrectomy for clinically serosa-positive gastric cancer (JACCRO GC-01 study)Eur J SurgOncol 2010 36(6):546-51. [Google Scholar]
[21]. Taiho Pharma USA. Phase 2 Study of S-1 in Combination with Cisplatin as 1st line therapy in advanced non-small cell lung cancer. Retrieved from http://clinicaltrials.gov/ct2/show/NCT00651833 on March 31, 2008 [Google Scholar]
[22]. Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, TS-1 advanced gastric cancer (AGC) clinical trial group. Randomized phase III study of S-1 alone versus S-1 + cisplatin in the treatment for advanced gastric cancer (The SPIRITS trial) SPIRITS: S-1 plus cisplatin vs S-1 in RCT in the treatment for stomach cancerJournal of Clinical Oncology 2007 25:18S [Google Scholar]
[23]. Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trialLancet Oncol 2008 9(3):215-21. [Google Scholar]
[24]. Nakamura K, Tahara M, Kiyota N, Hayashi R, Akimoto T, Fukuda H, Phase II trial of concurrent chemoradiotherapy with S-1 plus cisplatin in patients with unresectable locally advanced squamous cell carcinoma of the head and neck: Japan clinical oncology group study (JCOG0706)J ClinOncol 2009 39(7):460-63. [Google Scholar]
[25]. Ozawa Y, Inui N, Naitoh T, Yasuda K, Nagayama M, Shirai T, Phase II study of combination chemotherapy with S-1 and weekly cisplatin in patients with previously untreated advanced non-small cell lung cancerLung Cancer 2009 63(1):68-71. [Google Scholar]
[26]. Inokuchi M, Yamashita T, Yamada H, Kojima K, Ichikawa W, Nihei Z, Phase I/II study of S-1 combined with irinotecan for metastatic advanced gastric cancerBr J Cancer 2006 94(8):1130-35. [Google Scholar]
[27]. Ichikawa W, Takahashi T, Suto K, Yamashita T, Nihei Z, Shirota Y, Thymidylate synthase predictive power is overcome by irinotecan combination therapy with S-1 for gastric cancerBr J Cancer 2004 91(7):1245-50. [Google Scholar]
[28]. Nakajo A, Hokita S, Ishigami S, Miyazono F, Etoh T, Hamanoue M, A multicenter phase II study of biweekly paclitaxel and S-1 combination chemotherapy for unresectable or recurrent gastric cancerCancer Chemother Pharmacol 2008 62(6):1103-09. [Google Scholar]
[29]. Kazuaki T, Noriaki T, Hideki Y, Kazuhiro Y, Hideki O, Combination therapy with docetaxel and S-1 as a first-line treatment in patients with advanced or recurrent gastric cancer: a retrospective analysisWorld J Surg Oncol 2010 [Google Scholar]
[30]. Peters GJ, Noordhuis P, Van Kuilenburg AB, Schornagel JH, Gall H, Turner SL, Pharmacokinetics of S-1, an oral formulation of ftorafur, oxonicacidand 5-chloro-2,4-dihydroxypyridine (molar ratio 1:0.4:1) in patients with solid tumoursCancer Chemother Pharmacol 2003 52:1-12. [Google Scholar]
[31]. Komatsu T, Shimada N, Nagayama S, Yokoi T, Involvement of microsomal cytochrome P450 and cytosolic thymidine phosphorylase in 5-fluorouracil formation from tegafur in human liverClin Cancer Res 2001 7:675-81. [Google Scholar]
[32]. Van G, Schornagel JH, Gall H, Noordhuis DVMP, Turner SL, Swart MS, Phase I clinical and pharmacokinetic study of oral S-1 in patients with advanced solid tumoursJ Clin Oncol 2000 18:2772-79. [Google Scholar]
[33]. Takechi T, Uchida J, Mita A, Toko K, Takeda S, Unemi N, Antitumour activity and low intestinal toxicity of S-1, a new formulation of oral tegafur, in experimental tumour models in ratsCancer Chemother Pharmacol 1997 39:205-11. [Google Scholar]
[34]. Peters GJ, Kohne CH, Fluoropyrimidines as antifolate drugs. In: Jackman AL (ed.)Antifolate drugs in cancer therapy 1999 TotowaHumana Press:101-45. [Google Scholar]
[35]. Borner MM, Wit R, Caponigro F, Comella Greim G, Peters GJ, Patient preference and pharmacokinetics of oral modulated UFT versus intravenous fluorouracil and leucovorin: a randomised crossover trial in advanced colorectal cancerEur J Cancer 2002 38:349-58. [Google Scholar]
[36]. Humerickhouse RA, Haraf DM, Brockstein B, Stenson KM, Sulzen L, Ratain MJ, Phase I study of eniluracil, a dihydropyrimidine dehydrogenase inactivator, and oral 5-fluorouracil with radiation therapy in patients with recurrent or advanced head and neck cancerClin Cancer Res 1999 5:291-98. [Google Scholar]