Congenital Cystic Adenomatoid Malformation (CCAM) of the lung is a rare developmental abnormality with incidence rate of 1 in 11000 to 1 in 35000 live birth [1,2]. It happens due to bronchopulmonary foregut malformations that are probably caused by an arrest in lung development between 4th and 7th week of fetal life [1–3]. It does not have any genetic predisposition, gender predilection [4]. It is not affected by maternal factors such as race, age or environmental exposures [4].
There are reports showing that CCAM can transform into malignancies, such as bronchioloalveolar carcinoma [5], adenocarcinoma in aged [6], and pleuropulmonary blastoma [7]. Surgical removal of the affected lobe of the lung is the best treatment for CCAM. It also prevents future complications like recurrent infections and potential risk of malignant transformation [8–10]. CCAM is an uncommon congenital disease with limited data available in the form of case reports. It is important to identify such cases early so that early rectifying procedure can be attempted. Being a tertiary care hospital, our’s is a referral centre for treating such cases. This study was undertaken to present the experience of a tertiary care hospital in treating CCAM. The primary objective of the present study was to assess the clinical and radiological profile of CCAM in a tertiary care hospital. Secondary objectives were to study management and outcome of surgical intervention in case of CCAM.
Materials and Methods
The study was conducted at tertiary care academic institute. It was retrospective study conducted at tertiary care teaching institute. Patients’ data were collected from electronic medical records. All patients with either prenatal diagnosis of CCAM on antenatal ultrasound or patients having High Resolution Computed Tomography (HRCT) features suggestive of CCAM between 2006 to 2013 were included in the study. Other mimicking conditions like congenital lobar emphysema, bronchogenic cyst, bronchopulmonary sequestration, prior infection with pneumatocele and congenital diaphragmatic hernia were excluded.
A clinical questionnaire was prepared to retrieve data. It included age, gender, type and site of CCAM, symptoms at time of presentation, surgery conducted, duration of hospital stay and post-operative complication. Breathing difficulty or sucking difficulty was taken as equal. Acute respiratory distress was defined as hypoxic child with SPO2 <90% requiring supplemental oxygen. All patients had chest radiograph as initial investigation if there was high clinical radiological suspicion for CCAM, patients were subjected to CT chest with HRCT. Paediatric surgeon assessed the patient for feasibility of surgery and eligible patients were operated. Standard procedure was followed for surgery and patients were subjected to open thoracotomy under general anaesthesia. The affected lobes were removed and specimens were sent for histopathological analysis.
All included patients were followed up prospectively to find out about their current level of health. Via telephonic interview they were asked about their overall growth, quality of life, activity, rate of respiratory infections and requirement of hospital admission.
Statistical Analysis
Results are expressed as Mean±SD. The statistical analyses were done by SPSS version 11(SPSS inc. Chicago). Descriptive frequencies were expressed by mean±standard deviation.
Results
Total 15 patients with diagnosis of CCAM were included in the study. There were 8 (53.3%) male while the remaining 7 were female. Baseline characteristics of the study cohort are shown in [Table/Fig-1]. Five cases were detected on antenatal screening ultrasound while the rest were post-natal diagnosis. Three patients were also found to have other co-existing congenital abnormality: 2 with atrial septal defect and one with patent ductus arteriosus.
Baseline demographics of study population.
| Character | Value(n=15) |
|---|
| Age, year, median(range) | 0.3 (0.10-24) |
| Gender:
• Male
• Female | 8(53.3%)7(46.7%) |
| Symptoms at time of presentation
• Cough
• Breathing difficulty
• Fever
• ARDS
• Failure to thrive
• Recurrent pneumonia | 13(86.7%)11(73.3%)9(60%)6(40%)4(26.7%)4(26.7%) |
| Antenatal detection | 5(33.33%) |
| Other congenital abnormalities
• Atrial septal defect
• Patent ductus arteriosus | 2(13.3%)1(6.66%) |
The most common presentation was cough 13 (86%), breathing difficulty 11 (73%), fever 9 (60%), recurrent pneumonia 4 (26%), acute respiratory distress (hypoxia requiring supplemental oxygen) 6 (40%), one patient with abdominal distension and one presented with vomiting and seizure (others) 2 (12%).
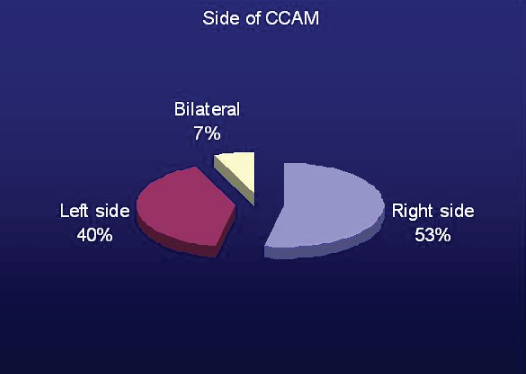
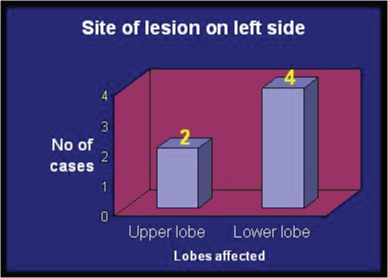
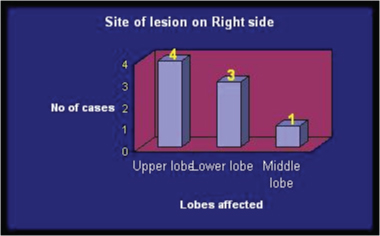
There were 8 case of right sided (4 Right Upper Lobe (RUL), 3 Right Lower Lobe (RLL), 1 Right Middle Lobe (RML), 6 cases of left sided CCAM (2 Left Upper Lobe (LUL), 4 Left Lower Lobe (LLL)), and 1 case of bilateral CCAM [Table/Fig-2,3 and 4]. Various radiology images of study patients are depicted in [Table/Fig-5].
Graph showing side of affected lung due to CCAM.
Graph showing lobar distribution on left side.
Graph showing lobar distribution on right side.
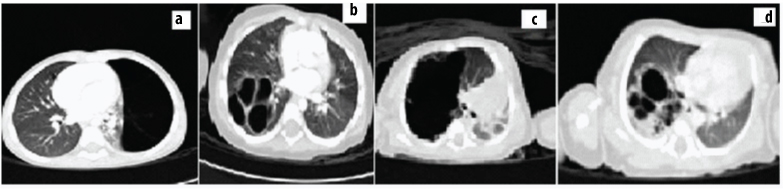
showing HRCT images showing various types of CCAM.
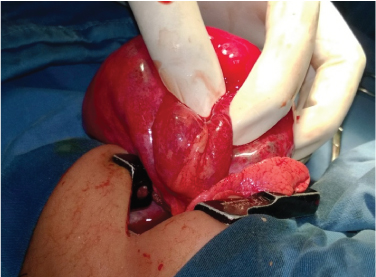
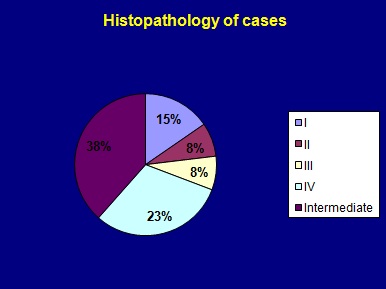
Thirteen patients were subjected to surgical management in the form of lobectomy. Gross appearance of CCAM is shown in [Table/Fig-6]. All were confirmed to have CCAM on histopathology [Table/Fig-7]. There were 2 cases of type I, one each of type II and III, 3 case of type 4 while 5 were intermediate type [Table/Fig-8] and one was reported as CCAM with haemorrhage. Patients were followed up in post-surgery period for post-operative complications. We did not observe any post-operative complications. There was no procedure related mortality and all were successfully discharged with median duration of stay 11±16 days. All patients had good post-operative recovery. One patient had bilateral disease and was not a candidate for surgery, so was managed conservatively and one patient was managed with medical management alone [Table/Fig-9].
Showing CCAM in post lobectomy gross specimen.
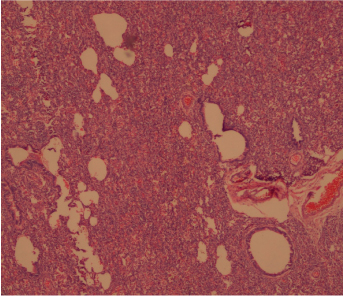
Histopathology picture showing congenital cystic adenomatoid malformation- Type 3. Bronchiolar structures are separated by densely packed irregular spaces lined by cuboidal epithelial cells, resembling fetal lung. {haemotoxylin and eosin (H&E) x100}.
Graph showing histopathology types of CCAM.
Primary and secondary end points.
| End point | Value n(%) |
|---|
| Primary end points
• Duration of hospital stay, days (median)
• Complications following the procedure | 11 (2 - 51)Nil |
| Secondary end pointsFollow-up
• Follow-up
• Procedure not done
• Lost for follow-up
• Overall growth
Normal
Average
Poor
• Respiratory Infections
< 1 in month
> 1 in month
• Hospital admission
< 3 in a year
> 3 in a year
• Activity capacity
Normal
Average
• Poor | 10 (66.6%)2(13.3%)3 (20%)7(70%)2(20%)1(10%)7(70%)3(30%)9(90%)1(10%)6(60%)4(40%)0 |
Included patients’ parents were contacted via telephone and enquired about child’s growth, activity score, and infection rate as well as need of hospitalization in post-surgical period. We could contact and arrange for follow-up in 10 patients. The information of follow-up is shown in [Table/Fig-9].
Discussion
In this series 15 cases of CCAM, a rare developmental, non-hereditary, hamartomatous abnormality of the lower respiratory tract were studied. Most of the population 12 (80%) were below 2 years of age. Male: Female were 1.14:1 and as described in literature we also did not find any significant gender predominance. Now-a-days, the majority of cases are identified by prenatal ultrasound examinations [9]. In our series there were 5 cases in which diagnosis was made on antenatal ultrasound. The median age of presentation in the study was 0.3±6 months.
As per literature, patients may present with symptoms, including cough, dyspnea, haemoptysis and respiratory distress, or remain asymptomatic [4]. The typical manifestations include progressive respiratory distress in the newborn and recurrent pulmonary infections in older children. In present series, cough and dyspnea were the commonest symptoms at time of presentation. However, there were six patients who presented with acute respiratory distress.
Recurrent or non-responding pulmonary infection is the most frequent complication of CCAM followed by haemopneumothorax, haemoptysis, and chronic cough [1,2].
Surgical resection of the affected part of the lung is the treatment of choice and this prevents recurrent infections and malignant transformation. Present study showed that lobectomy was a procedure with few complications [8]. It required a short hospital stay with mean duration of hospital stay of 11 days.
Its occurrence is sporadic and is not related to genetic predisposition, gender predilection, or maternal factors such as race, age, or environmental exposures. Stocker et al., suggested a new name, congenital pulmonary airway malformation and classified CCAM into five types based on the site of the defect in the tracheobronchial tree [2]. CCAM type 0 originates from tracheal or bronchial tissue, type 1 originates from distal bronchi or proximal bronchioles, types 2 and 3 have an acinar origin; type 4 have an acinar-alveolar origin.
Type 0 is the very uncommon, comprising 1 to 3% of cases. It originates from tracheal or bronchial tissue [11]. We did not have any case having type 0 CCAM.
Type 1 is the most common form of CCAM, comprising 60 to 70% [12]. In present study 15.4% of cases were of type I CCAM. The clinical presentation of type 1 of CCAM depends primarily on the size of the cysts. Larger cysts compressing the normal adjacent lung may be detected on prenatal ultrasound and can cause respiratory distress in the neonate, mediastinal shift to the contra-lateral side and flattening of the ipsilateral diaphragm. All two cases in our series had cough and breathing difficulty.
Type II lesions comprise 15 to 20% of CCAMs as per literature. They consist of multiple cysts 0.5 to 2 cm in diameter; resemble dilated terminal bronchioles lined with ciliated cuboidal or columnar epithelium. In present series there were 7.8% cases of CCAM type II. Extralobar pulmonary sequestrations may have a similar appearance, but unlike type II CCAM, these have a systemic blood supply. Type II CCAM is associated with other congenital abnormalities like esophageal atresia, tracheoesophageal fistula [13], bilateral renal agenesis or dysgenesis [14], intestinal atresia, other pulmonary malformations, and diaphragmatic, cardiac, central nervous system, and bony anomalies in about 60% of cases [2,11–13]. In our series the patient having CCAM type II also had Arterial Septal Defect (ASD). This association with other congenital malformations suggests that insults resulting in type 2 CCAMs occur during the third week of gestation. In many cases, the associated anomalies are the presenting feature, and the CCAM is detected as a secondary finding.
Incidence of Type III CCAM is 5 to 10%. In our study it was 7.8% which correlates with reported incidence. They have an acinar origin and are often very large involving an entire lobe or several lobes. Affected infants present in utero or at birth, with severe respiratory distress or death in the neonatal period [12]. Our patient reported at age of 4 months.
Type IV lesions comprise 10 to 15% of CCAMs [10]. In present series we found 23% cases of CCAM. Type IV CCAMs may present at birth or in childhood, often with tension pneumothorax or infection, but they may also be identified as incidental findings in asymptomatic patients. The time of presentation was 0.4 months – 3 year. The presenting symptoms were cough, fever, dyspnea, and one presented with recurrent lung infections. As type IV CCAM has been associated with the development of malignancies, such as bronchioloalveolar carcinoma, adenocarcinoma in aged, and pleuropulmonary blastoma, we feel surgery was treatment of choice in majority of our patients.
There were 5 cases of intermediate type. We did not find any significant post-operative complications and all patients had good post-operative recovery.
In present series, maximum number of patients had lobectomy. As per literature, after lobectomy usually the remaining lung expands well and grows so the pulmonary function test returns to normal [12,13].
In present study, follow-up was done of 10 cases out of 13 operated cases and their overall growth was assessed. Total 70% patients had normal overall growth and less than 1 episode of respiratory tract infection per month. Ninety percentage patients required less than 3 times hospital admission per year. However, we were not able to do pulmonary function test for any of the patients considering their young age.
English literature has multiple single case reports of CCAM with or without infections from India. One previous study from India included all kind of pulmonary malformation and was not specific for CCAM [14]. On literature search, one similar study from Argentina was found comprising 172 cases [15]. Comparison of our study with quoted study is shown in [Table/Fig-10].
Comparison of present study with previously published largest series.
| Variable | Presentstudy*(n=15) | Giubergia Vet al., 2012Argentina(n=172) [15] |
|---|
| Age at presentation | 0.3±6 months | 0.1-48 months |
| Male | 53.3 | 50.4 |
| Prenatal diagnosis | 33.33 | 14.53% |
| Clinical presentation
• Recurrent infection
• Respiratory distress
• Pleural effusion
• Asymptomatic | 26.740013.3 | 40%49%8.210 |
| Surgery
• Lobectomy/ Bilobectomy
• Pneumonectomy | 86.660 | 98.81.2 |
| Other congenital anomalies Cardiac | 20 | 10.5 |
| Lobar distribution of CCAM
• RUL
• RML
• RLL
• Lingular lobe
• Left LL
• Bilateral | 26.76.62013.326.76.66 | 22%12%38%4%24%0.58 |
| Type of CCAM
• 0
• 1
• II
• III
• IV
• Intermediate | 015.47.87.82338.4 | 0.5870240.5845.23 |
Limitation
Present study was retrospective in nature. The study numbers was also very small due to rarity of the disease. It was done at tertiary care hospital and so may not be applicable to general population as such. The study can be used as pilot study for future larger multi center trial.
Conclusion
CCAM is associated with significant morbidity if kept unattended. Removal of affected lobe is treatment of choice for CCAM. We found lobectomy a safe procedure with no significant post-operative complications and good post-operative recovery. Almost 70% patients who underwent surgery had normal growth, physical activity and lesser respiratory infections at 3 years interval.