Case Report
Case 1
A 10-year-old female presented with high grade fever on and off since 2 months and left sided hemiparesis for last 3 days before admission. Physical examination showed pallor and hepatomegaly (2cm below right costal margin). No history of bleeding, rashes, double vision, lymphadenopathy or splenomegaly was noted.
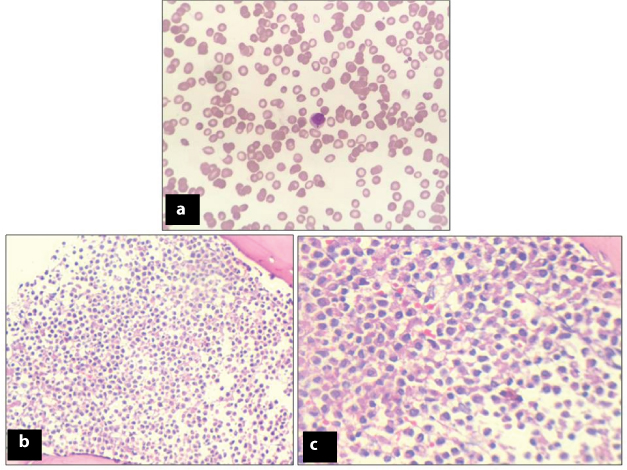
Laboratory investigations revealed haemoglobin of 3.2-g/dl, total leukocyte count of 2400/μl with differential count being polymorphs-54%, lymphocytes-45%, eosinophil-01%, and platelets-30,000/μl. Peripheral smear showed pancytopenia [Table/Fig-1a]. Red blood cells were normocytic normochromic and white blood cells showed leucopenia, however, no blasts were seen. Platelets were markedly reduced.
(a) Peripheral smear showing pancytopenia (Giemsa, X100). (b&c): Photomicrograph of Bone marrow biopsy showing leukemic promyelocytes (1b-H&E: X100 and 1c-H&E: X200).
Kidney and liver function tests were within normal limits. CT head showed a lesion in right temporo-parieto-occipital region, suggestive of infarction.
Bone Marrow Aspiration (BMA) smears were haemodilute, however, imprints were cellular showing a marked increase in leukemic promyelocytes constituting 77% of all nucleated cells [Table/Fig-1b,c]. Many Auer rods and faggots were also seen. Blasts constituted 7-8% of all nucleated cells. Blasts were intermediate to large in size with high nuclear-cytoplasmic ratio, opened up chromatin and scanty to appreciable amount of cytoplasm containing granules. Bone Marrow Biopsy (BMB) revealed cellular marrow spaces showing marked increase in leukemic promyelocytes and scattered blast cells with marked paucity of normal haematopoietic elements [Table/Fig-1b,c].
Peripheral blood, bone marrow aspirate & biopsy were suggestive of Acute promyelocytic Leukaemia (FAB subtype AML-M3). The patient was started on Tretinoin (All-trans-retinoic acid) and given other supportive management. However, the patient succumbed after two months of follow-up. The prognosis of such patient is poor as detailed in the discussion part below. In the case reported here, the patient had presented with cerebral infarction before initiation of any treatment, which makes it a very rare presentation.
Case 2
A 40-year-old female presented with complaints of low grade intermittent fever for last 2 months along with bone pain, decreased appetite and weight loss. General examination showed pulse rate: 80/min, blood pressure: 120/70 mm Hg, poor oral hygiene. No lymphadenopathy, pallor, icterus, cyanosis or oedema was seen. Ultrasound of abdomen revealed hepatosplenomegaly and periportal lymphadenopathy.
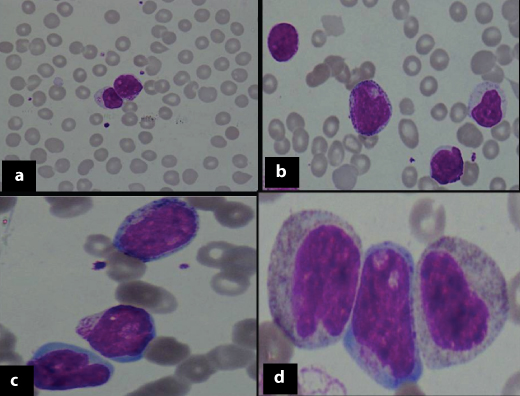
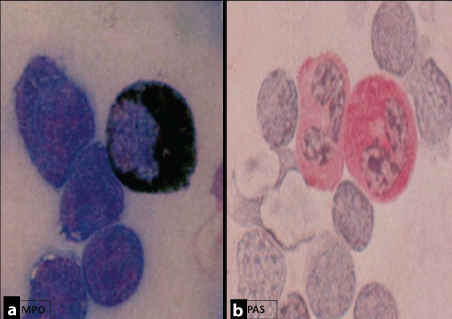
Laboratory investigations showed haemoglobin of 9.7 gm/dl, high total leukocyte count of 60,000/μl, platelet count of 1.8 lac/μl, haematocrit-29.7%, Mean Corpuscular Volume (MCV):92.2fl, Mean Corpuscular Hemoglobin (MCH):31.1pg, Mean Corpuscular Hemoglobin Concentration (MCHC): 33.7gm/dl. Peripheral smear showed 48% blasts, which were intermediate to large in size with high nuclear-cytoplasmic ratio, evident nucleoli, and moderate amount of cytoplasm with granules in 32% blasts [Table/Fig-2a-d]. Blood picture was normocytic normochromic. However, cytochemistry was inconclusive [Table/Fig-3a,b].
Atypical lymphoid cells and Blasts showing cytoplasmic granules (Giemsa stain, 2a: X100, 2b: X200, 2c: X400 and 2d: X1000, respectively).
Cytochemistry [Myeloperoxidase (a, X400) & PAS (b, X400)]: Negative in blast cells.
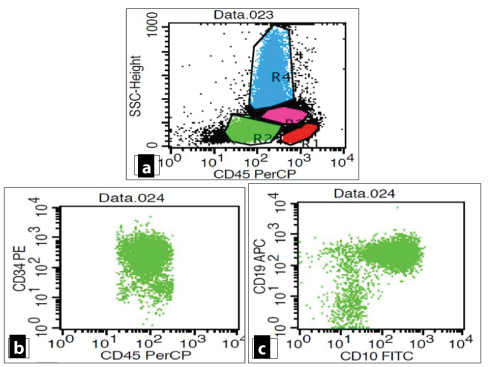
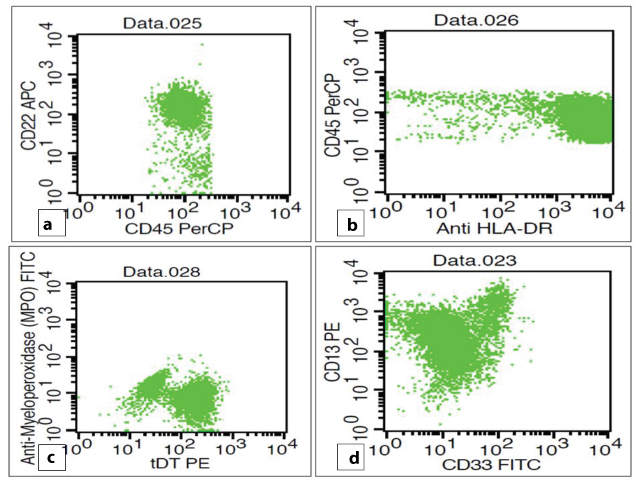
Flowcytometric analysis of peripheral blood revealed an abnormal cluster on CD45/SSC analysis, constituting 32% of total acquired events. These cells showed dim CD45 expression and low to intermediate FSC [Table/Fig-4a]. Bright positivity was noted for CD10, CD19, tDT, HLA-DR. However, CD22 and CD34 showed moderate expression [Table/Fig-4b,c and 5a-c]. Aberrant expression of CD13 (dim to moderate) was also noted [Table/Fig-5d].
a: CD45/SSC scattergram revealed an abnormal cluster showing dim CD45 expression (blast cells). b,c: Blasts showing moderate expression of CD34 with co-expression of CD10 and CD 19.
Blasts showing positivity for CD22 (moderate), HLA-DR (bright), tDT (dim to moderate) along with an aberrant expression of CD13 (dim to moderate).
Based on peripheral blood cytomorphology & flowcytometric immunophenotypic analysis, a diagnosis of “granular B cell acute lymphoblastic leukaemia with aberrant CD13 expression” was made. The patient was given supportive management and multidrug ALL protocol following which the patient had achieved initial complete remission.
Case 3
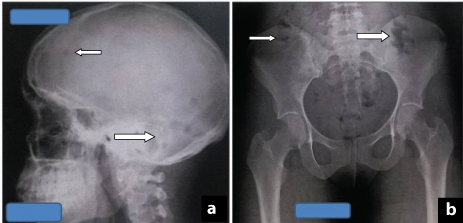
An 18-year-old male presented with multisystem involvement, with complaints of fever, vomiting, cough and swelling of feet. The patient had osteolytic lesions in skull and skeletal survey (pelvis) [Table/Fig-6 a,b], hypercalcaemia, decreased urinary output, haematuria, renal failure and skin lesions. Skin biopsy showed evidence of calcinosis cutis. Clinically, the differential diagnoses made were Multiple myeloma, Langerhans cell histiocytosis, T cell Acute lymphoblastic lymphoma.
(a) X-ray skull showing multiple osteolytic lesion. (b) X-ray pelvis showing multiple lytic lesions.
The patient was a known case of seizure disorder (Cysticercosis) and was on phenytoin for a year. Physical examination showed pallor and pedal oedema. Chest examination revealed bilateral crepts. Per abdomen showed hepatomegaly, 2cm below right costal margin. Bilateral plantar reflexes were decreased. Patient had received one unit of blood transfusion and underwent dialysis twice.
Chest X ray was found to be within normal limits. X-ray skull and skeletal survey (pelvis) showed multiple osteolytic lesions [Table/Fig-6a,b]. Ultrasound abdomen revealed hepatomegaly with evidence of chronic kidney disease.
Laboratory investigations showed haemoglobin of 7.5gm/dl, increased total leukocytic count of 30,000/μL and differential count showing polymorphs-54%, lymphocytes-06%, eosinophil-01%, blast cells-32%, myelocyte-07% and ESR of 35mm at the end of 1st hour. Biochemical laboratory values showed deranged kidney function tests with urea 192mg/dl (15-45mg/dl), creatinine- 2.9 mg/dl (0.6-1.2 mg/dl) and hypercalcaemia with calcium levels of 16mg/dl (8.5-10.2 mg/dl). However, liver function tests and serum electrolytes were within normal limits. No monoclonal band (M-band) was noted on serum protein electrophoresis. Peripheral smear showed leucocytosis. Blast cells constituted 30% of WBCs and were small to intermediate in size, having high nuclear-cytoplasmic ratio, round to clefted/ indented nuclei, some showing 1-2 nucleoli. A shift to left was seen, however, platelets were found to be within normal limits. Medical management was given for controlling hypercalcemia.
Bone marrow aspiration was repeated as the initial bone marrow aspirate was a dry tap. The smears were markedly cellular with presence of blast cells constituting 85% of all nucleated cells. Bone marrow biopsy showed almost complete replacement of normal haematopoietic elements by blasts. Based on peripheral blood and bone marrow aspiration findings, a diagnosis of acute Leukaemia was made and Flowcytometry (FCM) was suggested for further characterisation.
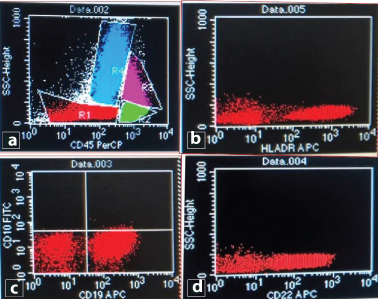
Flowcytometric analysis showed an abnormal cluster on CD45/SSC analysis, constituting 16 % of total acquired events. These cells showed dim CD45 expression and low to intermediate FSC. A moderate positivity of CD19 was noted along with moderate to bright expression of CD22 and HLA-DR [Table/Fig-7a-d]. Rest of the antibodies used in panel showed negative expression (CD13, CD33, CD117, CD34, CD10, CD5, CD7, CD14, CD64, MPO, tDT). Finally, a diagnosis of B cell-acute lymphoblastic leukemia was made based on peripheral blood, bone marrow aspiration findings and immunophenotypic analysis. The clinical, radiological, biochemical profile along with peripheral blood, bone marrow findings and FCM immunophenotyping have been tabulated [Table/Fig-8]. The patient was started on ALL chemotherapy regimen and achieved complete initial remission and is still under strict follow-up.
(a) CD45/SSC scattergram revealed an abnormal cluster showing dim CD45 expression (blast cells). (b-d) Blast cells showing moderate expression of CD19 as well as moderate to bright positivity for CD22 and HLA-DR.
Brief summary of cases (clinico-haematological, biochemical and radiological findings).
| Clinical Features | Radiological Findings | Haematological findings and Biochemical findings | BMA/BMB | Flowcytometric Immunophenotypicanalysis | Rare findings of individual case |
|---|
| APML | 10 yrs/F High grade fever Pallor Hemiparesis | USG whole abdomen: Hepatomegaly NCCT head: Right temporo-parieto-occipital region infarct | Pancytopenia | Leukaemic promyelocytes(77%) Auer rods Faggots Blasts: 7-8% | ------ | APML presenting with hemiparesis with infarct in Right tempero-parieto-occipitalregion |
| Granular B-ALL | 40yrs/F Low grade Fever with bone pain Decreased appetite and weight loss with poor oral hygiene | USG whole abdomen: Hepatosplenomegaly and periportal lymphadenopathy | Leukocytosis with 48% blasts with granules in 32% blasts | ------ | 32% blasts of all acquired events with dim CD45 expression. Bright positivity f or CD10, CD19, tDT & HLA-DR. Moderate expressionof CD22 & CD34 Aberrant expressionof CD13 (dim to moderate). | Granular B-ALL in adult female with moderate expression of CD22 & CD34 and aberrant expression of CD13. |
| B-ALL | 18yrs/M Multisystemic involvement fever, cough, vomiting Swelling in feet decreased urinary output, haematuria and renal failure skin disease | CXR: Normal X-Ray skull: Multiple osteolytic lesions USG whole abdomen: Hepatomegaly with e/o chronickidney disease | P/S: Leukocytosis with 30% blasts serum Protein electrophoresis: negative for M-band. Kidney function test deranged. Serum calcium elevated | Initial BMA: dry tap. 2nd BMA: Blasts 85% BMB: Complete replacement of haematopoietic elements by blasts | Haemodilute specimen containing 16% blasts of all acquired events showing dim CD45 expression. Moderate to bright positivity for CD22 & HLA-DR along with moderate expression of CD19 | B-ALL patient nitially presenting with hypercalcaemia and multiple osteolyticlesions on X-ray skull and pelvis. Flowcytometric analysis: CD19, CD22 and HLA-DR positivity. |
APML: Acute promyelocytic leukemia, ALL: Acute lymphoblastic leukemia, M:Male, F: Female, USG: Ultrasound, NCCT: Non-contrast computed tomography, CXR: Chest X-Ray, BMA/BMB: Bone marrow aspiration & biopsy.
Discussion
Acute Promyelocytic Leukaemia (APML) is a subtype of acute myelogenous leukemia characterized by abnormal hypergranular promyelocytes, auer rods and faggots in the bone marrow and peripheral blood. In APML, morbidity and mortality frequently results from the complications associated with bleeding and infections dominating the clinical picture, thereby overshadowing the rare thrombotic events like cerebral infarction and acute myocardial infarction [1,2]. This malignant haematologic disorder is a distinct subtype of acute myelogenous Leukaemia, identified by the French-American-British classification as AML-M3 and is cytogenetically characterized by the balanced and reciprocal translocation between chromosomes 15 and 17 with association of life threatening coagulopathy [1].
The role of coagulopathy has been proved in the causation of Disseminated Intravascular Coagulation (DIC), which includes hypercoagulable state and haemorrhage. The pathogenesis of coagulopathy in APML results from an interplay of three main factors [3]. Activation of the extrinsic pathway occurs following release of tissue factor and cancer pro-coagulants from malignant cells [3]. There is an increased annexin II receptor expression on the surface of the leukemic promyelocytes. Annexin II receptor binds plasminogen and tissue plasminogen activator which increases plasmin formation, thereby leading to increased fibrinolysis. Bleeding manifestations are seen because of increased proteolysis [3].
In the first case, the patient had presented with cerebral infarct before initiation of any treatment which makes it a very rare presentation for APML patient. The thrombotic episode can involve any organ [3]. There are case reports of patients presenting with acute myocardial infarction and ischemic stroke [3,4]. Usually, this kind of presentation is noted in adult patients with APML, however, in the present case infarction was noted in a 10-year-old female child, making it a rare presentation [2]. The patients presenting with thrombotic event portend a poor prognosis as seen in our patient [2]. Hence, such clinical presentation should always be kept in mind while evaluating peripheral smear or bone marrow.
Currently, APML is considered to be having the most favorable prognosis with correct diagnosis and early intervention while the scenario was totally different a few decades ago, when it was considered as the most fatal of all other subtypes [4]. As the onset of APML is associated with a significant and high mortality, immediate and correct treatment is required for the patients. Pancytopenia is commonly associated with haemorrhagic events [5]. The unusual feature in the first case is the presence of thrombotic cerebral infarction as the initial manifestation.
Granules in the lymphoblasts are a relatively uncommon morphologic finding, as noted in the second case presented here. Acute lymphoblastic leukemia patient rarely present with numerous cytoplasmic granules in blasts and this may lead to a wrong diagnosis of acute myeloid leukemia [5,6]. These azurophilic granules in the blast cells of a leukemia patient are usually characteristic morphological finding of acute myeloid leukemia. These can be confirmed using cytochemical stain like myeloperoxidase and can be well differentiated from lymphoblastic leukemia. It has been more commonly noticed in children, though the clinical significance has not been explained. A very few case reports have mentioned its presence in adults too, as in the present case [5].
It has been shown in few studies that granular ALL patients have poor prognosis as compared to ALL whereas no such correlation was noted in various other studies [6,7]. The diagnostic criteria used for granular ALL is presence of more than 1% of lymphoblasts having at least three or more clearly defined azurophilic granules [8]. But usually, the majority of cases of granular ALL often show more than 5% of lymphoblasts with small numbers of distinctive azurophilic granules [8]. These granules may be positive for Sudan Black B (SBB) [8]. Such unusual positivity of SBB further adds to diagnostic difficulties. A correct diagnosis of granular ALL is better made using cytochemical stain like myeloperoxidase. However, at times cytochemistry may fail to provide conclusive opinion. In these cases, immunophenotypic analysis using a comprehensive antibody panel is of utmost importance for a definitive diagnosis. In the present case also, the above mentioned criteria was used to diagnose Granular B-ALL, however, cytochemistry was inconclusive and hence FCM immunophenotypic analysis was advised for a definitive opinion. These granules are thought to be the result of dysplastic organelle formation, fusion, or degeneration. Ultra structurally, these cytoplasmic granules resemble multivesicular bodies or Gall Bodies [8].
Very few case reports of adult granular ALL have been described in the literature and till date in no study, series of patients have been reported. A review of literature revealed that only 15 cases of adult granular ALL have been reported [5,7]. The age of diagnosis in these adult cases of granular ALL ranged from 20 to 58 years with a slight female predominance (M:F:2:3) [5,7]. These patients usually presented with heavily granulated lymphoblasts in the blastic population of the bone marrow aspirates (varying from 25 % to 95%). Though the phenomenon is rare, the overall prognosis appeared to be not so good. The average remission rate, following initial chemotherapy treatment, was about 50% [5–7]. Some patients relapsed after initial remission. Though childhood granular ALL does not bear any prognostic significance, it is important to diagnose this entity as it is a poor prognostic factor in adult patients.
Acute lymphoblastic leukaemia is the most common malignancy in children and the common presenting symptoms are pallor, hepatosplenomegaly, lymphadenopathy, fever, bone pain and bleeding manifestations. Hypercalcemia is potentially a life- threatening event and is associated with non-specific gastrointestinal symptoms like nausea, vomiting, anorexia, weight loss. This is more commonly seen in adult, rarer in children (0.4%-1.3%) [9]. Children with other malignancies like rhabdomyosarcoma, hepatoblastoma, Hodgkins disease, Non-Hodgkins lymphoma, brain tumours, etc can present with hypercalcemia, however this has rarely been associated with ALL. Considering this manifestation as initial presentation, a diagnosis of leukemia can be delayed leading to non-specific treatment [9,10].
In the third case, an 18 year-old-male presented with multisystem involvement as mentioned in the case report. Initial laboratory investigation revealed normal peripheral smear with shift to left. No abnormal cells were noted. Subsequent blood, bone marrow and flowcytometric findings confirmed it to be a case of B-ALL. In largest series reported from St Jude Children’s Hospital, 10 children with haematopoietic malignancy had ALL induced hypercalcemia and only 7 had hypercalcaemia at the diagnosis, as noticed in the present case [9].
The purpose of highlighting hypercalcaemia as the initial manifestation of childhood Leukaemia is to increase awareness among clinicians in this field. It is advisable to measure serum calcium in similar cases, as child with hypercalcaemia can present with non-specific symptoms, as noted in the present case. The key element of treatment that helps in quick correction of hypercalcaemia along with other modalities is prompt intervention for leukemia. The present patient also had skin lesions which on biopsy showed evidence of calcinosis cutis. Usually, T cell-ALL present with skin manifestations but in the present case this was seen associated with B-ALL [10].
Hann et al., and Jonsson et al., found a significant correlation between the severity of bone pain and number of bones with lytic lesions noted on X-rays [11,12]. The index case presented with multiple osteolytic lesion on skull X-ray, but with normal haematological values initially, which caused a delay in diagnosis. Very few reports of childhood ALL presenting with severe osteopenia and pathological fractures have been found in the literature [9–14].
The present case is brought into notice because of so many rare presentations like hypercalcemia, lytic bone lesions and calcinosis cutis which needed a thorough work up to rule out the other differential diagnoses.
Conclusion
Coagulopathy and thrombotic manifestations like cerebral infarct are rarely observed in young patients. It requires high index of clinical suspicion for prompt diagnosis as the presentation with thrombotic event portends a poor prognosis. Early intervention is life saving in these cases.
Granular blasts can morphologically mimic a myeloblast and these cases can be wrongly labelled as AML. Cytochemistry may also at times be not conclusive. Hence, FCM immunophenotyping is a pertinent diagnostic tool for a definitive diagnosis and further management.
The varied presentation in ALL can at times be quite misleading. In addition to the usual presentation like organomegaly, lymphadenopathy, bone pain, cytopenias, presence of rare and unusual manifestations like hypercalcaemia, lytic lesions and skin involvement should also be kept in mind and investigative modality should be planned accordingly to arrive at a conclusive diagnosis. This case series brings about many rare presentations of acute leukemias. Moreover, it also highlights the role of comprehensive work up to arrive at a definitive diagnosis so that optimal management can be offered to the patients.
[1]. Sharma JB, Gupta SK, Acute thrombotic occlusion of internal carotid artery: A rare neurological presentation of acute promyelocytic leukemiaNeurol India 2013 61:662-64. [Google Scholar]
[2]. Grisold W, Oberndorfer S, Struhal W, Stroke and Cancer: a reviewActa Neurol Scand 2009 119:1-16. [Google Scholar]
[3]. Kapoor R, Pati HP, Gupta SK, Gupta N, Acute Promyelocytic leukemia presenting as ischemic stroke in youngIndian Journal of Haematology and Blood Transfusion 2013 29(2):93-5. [Google Scholar]
[4]. Kwaan HC, Wang J, Boggio LN, Abnormalities in haemostasis in acute promyelocytic LeukaemiaHaematol Oncol 2002 20:33-41. [Google Scholar]
[5]. Fulcher JW, Allred TJ, Kulharya A, Satya Prakash KL, Seigler M, Neibarger D, Granular acute lymphoblastic Leukemia in adults: Report of a case and review of the literatureSouth Med J 2006 99:894-97. [Google Scholar]
[6]. Cerezo L, Shuster JJ, Pullen DJ, Brock B, Borowitz MJ, Falletta JM, Laboratory correlates and prognostic significance of granular acute lymphoblastic Leukemia in children: A Pediatric oncology group studyAm J Clin Pathol 1991 95:526-31. [Google Scholar]
[7]. Pitman SD, Huang Q, Granular acute lymphoblastic Leukemia: A case report and literature reviewAm J Haematol 2007 82:834-37. [Google Scholar]
[8]. Tembhare PR, Subramanian PG, Sehgal K, Yajamanam B, Kumar A, Gujral S, Hypergranular precursor B-cell acute lymphoblastic Leukemia in a 16-year old boyIndian J Pathol Microbiol 2009 52:421-23. [Google Scholar]
[9]. Amita T, Timothy C, Simon B, Hypercalcaemia in acute lymphoblastic Leukemia: an overviewJ Pediatr Haematol Oncol 2009 31(6):424-27. [Google Scholar]
[10]. Watson KMT, Mufti G, Salisbury JR, Vivier AWP, Creamer D, Spectrum of clinical presentation, treatment and prognosis in a series of eight patients with leukemia cutisClin Exp Dermatol 2006 31(2):218-21. [Google Scholar]
[11]. Hann IM, Gupta S, Palmer MK, Morris-Jones PH, The prognostic significance of radiological and symptomatic bone involvement in childhood acute lymphoblastic LeukemiaMed Pediatr Oncol 1979 6(1):51-5. [Google Scholar]
[12]. Jonsson OG, Sartain P, Ducore JM, Buchanan GR, Bone pain as an initial symptom of childhood acute lymphoblastic Leukaemia: association with nearly normal haematologic indicesJ Pediatr 1990 117(2):233-37. [Google Scholar]
[13]. Hafiz MG, Islam A, Extensive skeletal lesions in childhood acute lymphoblastic LeukemiaMymensingh Med J 2009 18(1):88-94. [Google Scholar]
[14]. Verma SP, Dubashi B, Basu D, Dutta TK, Kar R, A rare case of adult acute lymphoblastic leukemia presenting with paraparesis and multiple osteolytic lesionsIndian Journal of Haematology and Blood Transfusion 2014 30(1):24-26. [Google Scholar]