A Commonly Missed Well Known Entity- Acute Intermittent Porphyria: A Case Report
Smilu Mohanlal1, Radha Gulati Ghildiyal2, Alpana Kondekar3, Poonam Wade4, Richa Sinha5
1 Senior Resident, Department of Pediatrics, Topiwala National Medical College and BYL Nair CH. Hospital, Mumbai, Maharashtra, India.
2 Professor, Department of Pediatrics, Topiwala National Medical College and BYL Nair CH. Hospital, Mumbai, Maharashtra, India.
3 Associate Professor, Department of Pediatrics, Topiwala National Medical College and BYL Nair CH. Hospital, Mumbai, Maharashtra, India.
4 Associate Professor, Department of Pediatrics, Topiwala National Medical College and BYL Nair CH. Hospital, Mumbai, Maharashtra, India.
5 Resident, Department of Pediatrics, Topiwala National Medical College and BYL Nair CH. Hospital, Mumbai, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Smilu Mohanlal, Department of Pediatrics, 1ST Floor, College Building, Topiwala National Medical College and BYL Nair Hospital, Mumbai Central, Mumbai-400008, Maharashtra, India.
E-mail: drsmilu@gmail.com
Acute Intermittent Porphyria (AIP) usually presents with abdominal pain, peripheral neuropathy and psychiatric manifestations. Incidence of AIP being 5 in 1,00,000. We present a case of an 11-year-old male child with multiple cranial nerve involvement, quadriparesis, focal convulsions, hypertension, hyponatremia with history of recurrent abdominal pain. His complete haemogram, ultrasonography (USG) abdomen, renal function tests were normal, he was also evaluated for tuberculosis which was negative. On further evaluation Electroencephalography (EEG) was suggestive of a generalised seizure disorder, MRI Brain suggestive of Posterior Reversible Encephalopathy Syndrome (PRES), Electromyography revealed a sensory motor axonal polyneuropathy and urine UV fluoresence test was positive for porphobilinogen which clinched the diagnosis of AIP.
Abdominal pain, Posterior reversible encephalopathy syndrome, Seizures
Case Report
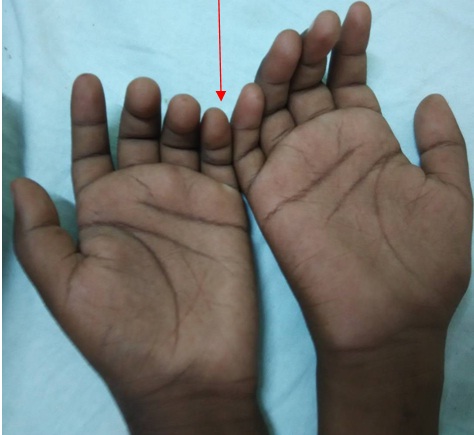
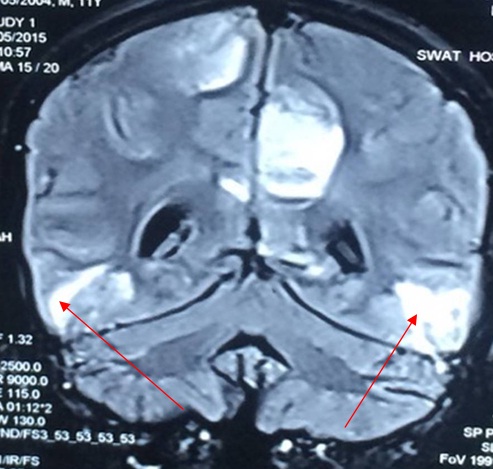
An 11-year-old boy presented with the history of intermittent abdominal pain and seizures of 4 months duration. His initial admission was in view of peri-umbilical abdominal pain and one episode of generalised tonic clonic convulsions without focal neurological deficits. His lumbar puncture analysis, Computed Tomography of brain, USG abdomen was normal. His EEG revealed generalised seizure activity. He was treated symptomatically and discharged on anticonvulsants (Tab. Levetiracetam and Tab. Sodium Valproate). He was re-admitted within a month with abdominal pain and acute onset quadriparesis. He was treated symptomatically and started empirically on antitubercular treatment. His neurological deficit remained the same but seizure frequency increased. After three weeks he presented again with altered sensorium, respiratory depression and increased frequency of focal convulsions for which he required ventilator support for 10 days, anticonvulsants and antibiotics. He improved and was weaned off from ventilator, managed symptomatically and discharged. All the previous three admissions were at a private hospital. He then presented to us after a fortnight with increased frequency of seizures, right sided facial palsy and peri-umbilical pain. On further enquiry, he had intermittent episodes of passing high coloured urine. On examination he had hypertension (B.P- 150/100 mmHg greater than his 95th centile), pallor and claw hand with thenar and hypothenar atrophy [Table/Fig-1]. His neurological examination revealed normal higher mental functions. Cranial nerve examination was suggestive of right Lower Motor Neuron (LMN) facial nerve palsy, IXth and Xth cranial nerve palsy. He had quadriparesis, hypotonia, areflexia with absent plantar response on motor system examination. He had no cerebellar signs and his sensory and autonomic nervous system examination were within normal limits. There was no local tenderness and no organomegaly on per abdominal examination. Rest of the systemic examination were within normal limits. His Complete Haemogram showed haemoglobin-11gm%, total counts- 7000 cells/cu.mm, platelets- 2.4 lac cells/cu.mm, blood urea nitrogen-17mg%, Serum Creatinine- 0.36mg%. His liver enzymes were altered: Aspartate Transaminase (AST)/Alanine Transaminase (ALT)- 119/162 IU which gradually improved to 60/82 respectively over a period of two weeks, total bilirubin/direct bilirubin- 1.0/0.5mg%, PT/INR-12secs/1.0, Total protein/serum.albumin-5.6/3.2gm%. Child also had persistent hyponatremia (115-120meq/litre) which responded to correction. Our differential diagnoses were periodic paralysis, chronic meningoencephalitis, (Acute Disseminated Encephalomyelitis) (ADEM), AIP and Guillain-Barre’s syndrome. His electromyography and nerve conduction studies revealed a sensory motor axonal polyneuropathy, MRI brain and spine showed bilateral symmetric vasogenic oedema mainly involving the subcortical white matter in the parieto-occipital, posterior temporal, and posterior frontal lobes as seen in PRES [Table/Fig-2]. His anti tubercular therapy was discontinued and he was treated symptomatically with anticonvulsants (Levetiracetam 20 mg/kg/day orally), sodium correction with 3% normal saline, antihypertensive (Labetalol 3mg/kg/hr), physiotherapy and nutritional rehabilitation. With the above symptomatology and investigations AIP was considered. His urine examination was performed which showed UV fluoresence positive for porphobilinogen. Parents were counselled about the acute attacks, precautionary measures were advised and confirmatory tests like DNA analysis were planned. He was discharged after 20 days of hospitalisation. On discharge his blood pressure was 100/76mm Hg (at the 50th centile), gag reflex was present, right sided facial weakness had improved and Serum sodium was 136 meq/dl. He was discharged on anti convulsants. On follow-up, the quadriparesis in our patient resolved in over 6 months, seizures were well controlled and he had no further attacks of abdominal pain.
Illustration of claw hand in our patient.
MRI brain showing bilateral symmetric vasogenic oedema mainly involving the subcortical white matter in the parieto-occipital region suggestive of posterior reversible encephalopathy.
Discussion
Our patient had variable presentation with multisystem involvement such as quadriparesis, right LMN facial nerve palsy, hypertension, Syndrome of Inappropriate Anti diuretic Hormone (SIADH), the constellation of such manifestations together were unexplained by the differentials considered. The neurological presentations were given importance over recurrent abdominal pain which led to the initial dilemma in the diagnosis. The association of episodic abdominal pain with dark urine supported by a UV fluorescence positive for porphobilinogens confirmed the diagnosis of AIP.
The key differential diagnoses in our case were chronic meningoencephalitis, ADEM, Guillian-Barre’s syndrome and periodic paralysis. The progression of symptoms despite appropriate antibacterial and anti tubercular therapy ruled out chronic meningoencephalitis, MRI brain and spine findings helped us to rule out ADEM and Guillain-Barre’s Syndrome (GBS). The constellation of features like intermittent abdominal pain, neurological findings, SIADH, hypertension supported by urinary porphobilinogen positivity test helped us arrive at the diagnosis of AIP in the absence of cutaneous manifestations.
The key enzyme in heme synthesis is porphobilinogen deaminase, deficiency of which leads to AIP [1,2]. Acute attacks can be triggered by starvation, drugs such as rifampin, barbiturates, hydantoin, sulphonamides and endogenous steroid hormones and other intercurrent illnesses or stress [3]. The classical triad of AIP is seen in 50% of the patients which include intermittent abdominal pain, neuropathy and mental changes. AIP with renal failure can present with skin blisters [4]. Diagnosis of porphyria is based upon the key clinical and laboratory features like recurrent abdominal pain with neurological manifestations like seizures, hypertensive crisis, dysautonomia supported by elevated urinary porphobilinogens, enzyme analysis or DNA mutation analysis [5].
Abdominal complaints like pain, vomiting, diarrhoea, distension constitutes 90% of the acute autonomic neuropathy. Peripheral neuropathy accounts for 10-40% of the episodes. Of the children presenting with acute neuropathy, 75% have cranial nerve involvement where the nerves involved predominantly are facial, vagus, trigeminal, occulomotar and accessory nerves. Encephalopathy manifesting as seizures constitutes 75% of the attacks. Complex partial seizures are the most common type. Others include generalised tonic clonic, myoclonic and absence seizures [6]. The acute attacks have been associated with hyponatremia secondary to SIADH caused by neuronal loss in supraoptic and paraventricular nuclei of hypothalamus or due to renal and gastrointestinal losses. Most of the cases with acute neuropathy resolve by itself. MRI brain in the early stages shows evidence of reversible cortical changes like PRES [7]. Less frequently MRI brain in AIP shows central pontine myelinosis [8]. PRES in a case of porphyria is not uncommon and is due to reversible cerebral vasospasm due to neurotoxic effects of porphyrins or a consequence of hypertensive encephalopathy. Other theories are due to the deficiency of nitric oxide, as nitric oxide synthase requires heme and heme is deficient during the acute attacks or due to cerebral auto-regulatory dysfunction [9–11]. PRES changes in MRI brain are self limiting and usually disappear with the patient requiring only symptomatic management [11].
Beta blockers are used for the management of hypertension secondary to sympathetic overactivity and antiepileptics preferred include gabapentin, vigabatrin and levetiracetam. Treatment is mainly supportive but patients with delirium and weakness need intensive care management. Severe attacks need treatment with intravenous heme which is given in the form of panhematin in the dose of 3mg/kg/day for 4-5 days [12]. Heme acts by reducing the activity of ALA synthetase which results in rapid reduction of hepatic overproduction of ALA and porphobilinogens.
The motor neuropathy seen in AIP improves over months though residual weakness may persist. Progression of the neuropathy involving the respiratory muscles and bulbar palsy are uncommon. Death is uncommon with appropriate treatment.
Conclusion
AIP can cause a diagnostic dilemma due to its varied manifestations. Thus, in a child with episodic acute abdominal pain with central nervous system manifestations, AIP should be considered as one of the differential diagnosis. PRES can be a neuro-radiological finding in a case of AIP.
[1]. Li M, Vizzard MA, Jaworski DM, Galbraith RA, The weight loss elicited by cobalt protoporphyrin is related to decreased activity of nitric oxide synthase in the hypothalamusJ Appl Physiol 2006 100:1983-91. [Google Scholar]
[2]. Nordmann Y, Puy H, Da Siva V, Simonin S, Robreau AM, Bonaiti C, Acute intermittent porphyria. The prevalence of mutations in the porphobilinogen deaminase gene in blood donors in FranceJ Intern Med 1997 242:213-17. [Google Scholar]
[3]. Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Recommendations for the diagnosis and treatment of the acute porphyriasAnn Intern Med 2005 142:439-50. [Google Scholar]
[4]. Sardh E, Andersson DE, Henrichson A, Harper P, Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimesCell Mol Biol 2009 55:66-71. [Google Scholar]
[5]. Pischik E, Kauppinen R, An update of clinical management of acute intermittent porphyriaAppl Clin Genet 2015 8:201-14. [Google Scholar]
[6]. Simon NG, Herkes GK, The neurological manifestations of the acute porphyriasJ Clin Neurosci 2011 18:1147-53. [Google Scholar]
[7]. Celik M, Forte H, Dalkilic T, Babacan G, MRI reveals reversible lesions resembling posterior reversible encephalopathy in porphyriaNeuroradiology 2002 44:839-41. [Google Scholar]
[8]. Susa S, Daimon M, Morita Y, Kitagawa M, Hirata A, Manaka H, Acute intermittent Porphyria with central pontine myelinolysis and cortical laminar necrosisNeuroradiology 1999 41:835-39. [Google Scholar]
[9]. Bartynski WS, Posterior reversible encephalopathy syndrome, part 1: controversies surrounding pathophysiology of vasogenic oedemaAJNR 2008 29:1043-49. [Google Scholar]
[10]. Kupferschmidt H, Bont A, Schnorf H, Landis T, Walter E, Peter J, Transient cortical blindness and bioccipital brain lesions in two patients with acute intermittent porphyriaAnn Intern Med 1995 123:598-600. [Google Scholar]
[11]. Kupferschmidt H, Bont A, Schnorf H, Landis T, Walter E, Peter J, Transient cortical blindness and bioccipital brain lesions in two patients with acute intermittent porphyriaAnn Intern Med 1995 123:598-600. [Google Scholar]
[12]. Besur S, Hou W, Schmeltzer P, Bonkovsky HL, Clinically important features of porphyrin and heme metabolism and the porphyriasMetabolites 2014 4:977-1006. [Google Scholar]