Amyloidosis is characterized by pathological deposition of abnormal protein aggregates in various tissues, AL protein being the commonest. ALECT 2 is the newest protein described, having a predisposition to affect the kidneys, sometimes the liver and rarely other organs. We present a case of renal amyloid ALECT 2 due to leucocyte cell derived chemotaxin 2, a novel amyloidogenic protein. The patient presented with mild proteinuria, scattered plasma cells on bone marrow examination and altered kappa/lambda ratio with associated cytogenetic abnormality of der7q add(7). It is essential to correctly type this protein and differentiate it from AL during diagnosis for appropriate and effective clinical management.
Leucocyte derived chemotaxin 2, AL Protein, der7q add(7), Renal amyloidosis
Case Report
A 52-year-old hypertensive male with Chronic Kidney Disease, Stage 3 (CKD 3) was admitted for evaluation of gradually worsening renal function. Drug history included angiotensin receptor and methylprednisone for intractable allergic manifestations including rhinorrhoea and pruritis since about a month. Examination revealed bilateral pitting pedal oedema, with a blood pressure 140/90mm Hg and no significant postural drop. He was found to have pallor, no palpable lymphadenopathy, no hepatospenomegaly and no neurological symptoms.
Serum creatinine was 2.1mg/dl, which went up from 1.6mg/dl over a two month period. Urine examination showed 1+ proteinuria. Ultrasonography of the abdomen revealed normal sized kidneys and no specific findings.
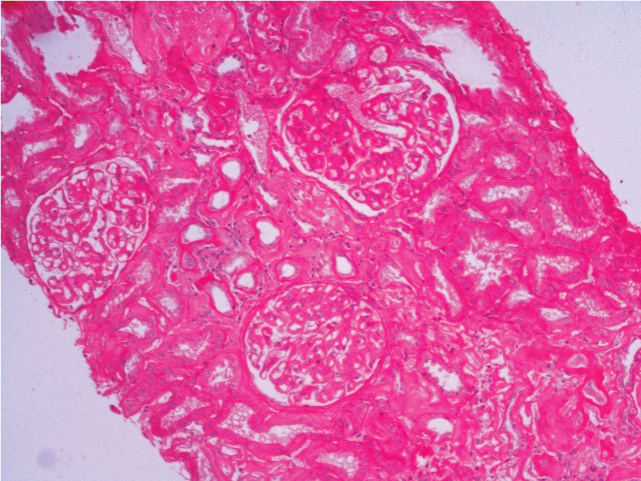
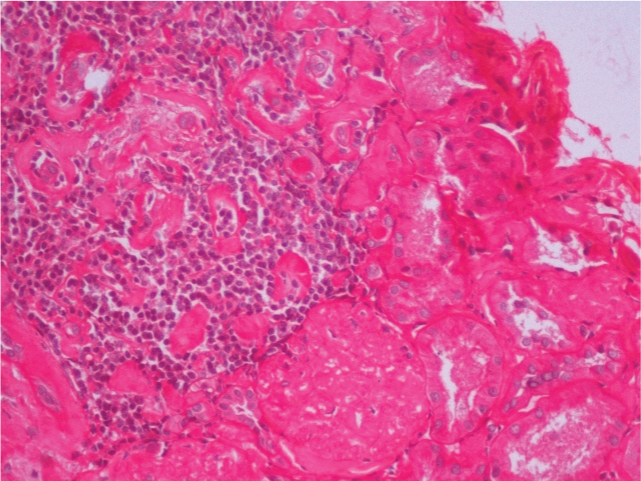
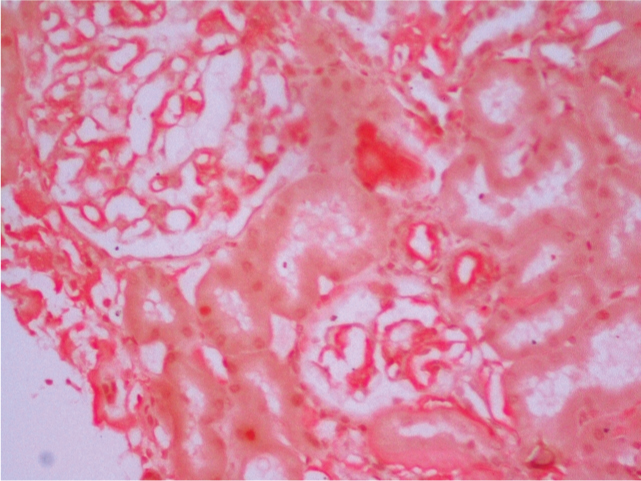
In view of his clinical profile of unexplained renal dysfunction and proteinuria, needle biopsy of the kidney was performed. Microscopic examination revealed kidney tissue containing glomeruli with patchy tubular atrophy and mild chronic inflammation [Table/Fig-1,2]. Deposition of Congophilic and eosinophilic material was noted in the glomeruli, blood vessels and interstitium [Table/Fig-3]. These deposits showed apple green birefringence under polarized light. Immunofluorescence showed absence of any IgG, IgM, IgA, C3 and C1q deposition. Hence, a diagnosis of AL Amyloidosis was made.
Light microscopy findings- Kidney tissue showing glomeruli and patchy tubular atrophy. (H&E, 10X).
Kidney tissue showing mild chronic interstitial inflammation. (H&E, 20X).
Deposition of Congophilic and eosinophilic material in the glomeruli, blood vessels and interstitium. (Congo red stain, 40X).
Further investigations were done to establish the cause of amyloidosis and to find out its association with any underlying clonal plasma cell disorder. Serum protein electrophoresis and Immunofixation were normal. Serum Beta 2 microglobulin was 3.5 mcg/ml with absent urinary Bence Jones protein. Serum free light chain assay revealed a Kappa: Lambda ratio of 1.865, which was normal as per his creatinine clearance. Bone marrow examination showed scattered plasma cells with no light chain restriction on immunohistochemistry. Cytogenetic study from the bone marrow showed 46,XY,der(7)add(7)(q34) pattern.
Paraffin embedded sections of the renal biopsy were sent for further typing of the amyloid protein. Liquid Chromatography tandem Mass Spectrometry (LC MS/MS) detected a peptide profile consistent with ALECT2 type amyloid deposition. These findings support the diagnosis of amyloidosis and indicate ALECT2 type amyloid deposition.
The patient improved symptomatically with conservative management without initiation of any chemotherapy. A follow up creatinine value of 1.3 mg/dl was documented over a 2-3 months period. He was advised close nephrological and haematological follow up with no active intervention at that time.
Discussion
Amyloid is defined as unique protein material deposited invivo extracellularly, presenting as homogeneous eosinophilic deposition on routine histology sections, distinguished from non-amyloid deposits by its characteristic ultrastructural fibrils and affinity to Congo red stain, showing green birefringence under polarized light. It is formed due to proteins misfolding into an insoluble beta-pleated sheet physical format. The clinical manifestations, prognosis and therapy greatly vary depending upon the specific type of amyloidosis [1]. However, limited treatment options are available.
A most recently identified novel type of amyloid is the leukocyte chemotactic factor 2 (ALECT2) protein. LECT2 was originally isolated and characterized from culture media of Phytohemagglutinin (PHA)-activated human T-cell leukemia cells as part of studies to identify chemotactic factors important in the pathophysiology of neoplasia, which happened to be amyloidogenic. The human LECT2 gene has been localized to chromosome 5 (5q31.1-q32) and consists of four exons and three introns [2].
LECT2 gene was discovered in 2008, and is currently being diagnosed in increasing number of cases. Larsen CP et al., analysed 44 cases of amyloidosis and identified ALECT2 as the second most common type of amyloid next to AL amyloidosis (83.3%) and was seen in about 10% of cases of amyloidosis [3]. Another study conducted by Theis et al., showed ALECT2 to be the third most common amyloid after AL and AA amyloid [4].
A large majority of the cases of ALECT2 have been reported from Southwestern United States, most of the patients being of Hispanic origin. Other significant populations were the Native Americans and Middle Eastern origin population [5,6]. An earlier description from India [7] was about a patient of the Punjabi descent, generally found in the north western part of this country. Our patient is an Indian Bengali, originating from the far eastern part of India. This shows that the presence of ALECT2 is not restricted to certain parts and may have a much wider presence than previously thought.
Initially thought of as a renal limited amyloidosis, now ALECT2 has been proved to be of more systemic distribution affecting the liver, spleen, colon and adrenal glands [2,8], commonly presenting with varying degrees of impaired kidney function. Proteinuria, as seen in our case is low grade in comparison to other forms of amyloidosis [8]. In a recent study, Said et al., evaluated 72 patients with renal ALECT2 amyloid. Proteinuria was not very characteristic and was absent in a third, whereas nephritic syndrome and haematuria were still rare [6]. Hypertension was present in 68.1% and ~10% presented with end stage renal failure requiring haemodialysis. Only about 10% of patients tested for monoclonal protein had a monoclonal gammopathy. Larsen et al showed that majority of these cases presented with chronic renal disease and interestingly 40% of patients did not have proteinuria. This lower grade proteinuria or its absence could be due to lack of glomerular involvement [5].
This case report addresses the possibility of primary amyloidosis being one of the under diagnosed causes of renal dysfunction. Such findings, in the setting of stable CKD, may be significant in the future in terms of determining management protocols and assessing the risks of developing a possible monoclonal gammopathy. However, more importantly it signifies the importance of typing the amyloid protein and to evaluate whether the amyloid related organ damage is due to AL or non-AL disease. In fact, it is imperative that no harm comes to the patient by administering unnecessarily toxic chemotherapy presuming the case to be of AL amyloid.
A rare association seen in this case is the cytogenetic abnormality detected with primary amyloidosis. der(7)add(7)(q34) chromosomal pattern on cytogenetics has not been documented in literature hitherto.
Few studies have shown a causal relationship of chromosome 7 with Systemic amyloidosis [9]. Although cytogenetic analysis of this nature in the plasma cells of patients with AL amyloidosis has not yet been reported, some studies have sought to determine if the bone marrow plasma cells of such patients have the same numerical chromosomal abnormalities as those reported in Multiple Myeloma (MM) and Monoclonal Gammopathy of Undetermined Significance (MGUS). Moreover, another study [10] has shown serum amyloid A gene complex to be proximal to chromosome 7 breakpoint in T(7;x). The frequent association of chromosome 7 abnormalities and systemic amyloidosis may prove to have diagnostic and prognostic significance in larger studies and reviews.
With scarcity of information, specific therapy for ALECT 2 is yet to be established. It seems that patients need to be individually assessed based on specific features and requirements, like kidney transplantation or haemodialysis [5].
Furthermore, the possibility of primary amyloidosis pre-empting the diagnosis of occult myeloma is well known. In this context, the findings of scattered plasma cells, Serum free light chain assay of raised kappa: lambda, and raised beta 2 microglobulin may well point to this direction.
Conclusion
To establish the causal relationship of chromosome 7 abnormalities with ALECT2 amyloid or myeloma is yet to be established and association with prognosis or treatment. With increasing number of cases with ALECT2 now being identified, it seems likely that specific guidelines in the management of ALECT2 amyloidosis will be formulated in the near future.
[1]. Leung N, Nasr SH, Sethi S, How I treat amyloidosis: the importance of accurate diagnosis and amyloid typingBlood 2013 120:3206-13. [Google Scholar]
[2]. Jiménez-Zepeda VH, Leung N, ALECT2, amyloidosis, renal dysfunctionRev Invest Clin 2014 66(4):269-73. [Google Scholar]
[3]. Larsen CP, Kossmann RJ, Beggs ML, Solomon A, Walker PD, Clinical, morphologic, and genetic features of renal leukocytechemotactic factor 2 amyloidosisKidney Int 2014 86(2):378-82. [Google Scholar]
[4]. Theis JDDS, Vrana JA, Mereuta O, Grogg K, Gertz M, Zeldendrust S, Proteome of amyloidosis: Mayo Clinic Experience in 4139 casesBlood 2013 2013:1900 [Google Scholar]
[5]. Vela-Ojeda J, García-Ruiz Esparza MA, Padilla-González Y, Sánchez-Cortes E, García-Chávez J, Montiel-Cervantes L, Multiple myeloma-associated amyloidosis is an independent high-risk prognostic factorAnn Haematol 2009 88(1):59-66. [Google Scholar]
[6]. Said SM, Sethi S, Valeri AM, Chang A, Nast CC, Krahl L, Characterization and outcomes of renal leukocyte chemotactic factor 2-associated amyloidosisKidney Int 2014 86(2):370-77. [Google Scholar]
[7]. Kulkarni UP, Valson A, Mathews V, Leukocyte derived chemotaxin 2 (ALECT2) amyloidosisMediterr J Hematol Infect Dis 2015 7(1):e2015043 [Google Scholar]
[8]. Murphy CL, Wang S, Kestler D, Larsen C, Benson D, Weiss DT, Leukocyte Chemotactic Factor 2 (LECT2)-associated renal amyloidosis: A Case SeriesAm J Kidney Dis 2010 56(6):1100-07. [Google Scholar]
[9]. Fonseca R, Ahmann GJ, Jalal SM, Dewald GW, Larson DR, Therneau TM, Chromosomal abnormalities in systemic amyloidosisBr J Haematol 1998 103(3):704-10. [Google Scholar]
[10]. Taylor BA, Rowe L, Genes for serum amyloid A proteins map to Chromosome 7 in the mouseMol Gen Genet 1984 195(3):491-99. [Google Scholar]