Macrophage Activation Syndrome (MAS) is a rare form of Haemophagocytic Lymphohistiocytosis (HLH) associated with rheumatologic conditions. It is characterised by haemophagocytosis and cytokine overproduction, resulting from the activation and uncontrolled proliferation of T lymphocytes and macrophages. The patient being reported is an elderly female with Rheumatoid Arthritis (RA), who presented with fever and was found to have MAS. She later developed Acute Respiratory Distress Syndrome (ARDS) and died. Such a scenario has rarely been reported.
Case Report
A 55-year-old female presented to emergency department with complaints of high grade continuous fever of eight days duration and was taking only paracetamol. She was on treatment for RA with methotrexate (10 mg once weekly), hydroxychloroquine (400 mg once daily) and folic acid, for the past five years. A Computed Tomography (CT) same of thorax was taken about six months ago; and was reported to show mild left sided Interstitial Lung Disease (ILD) (films not available), but was not on any medications or oxygen therapy. She did not have any other co-morbid conditions.
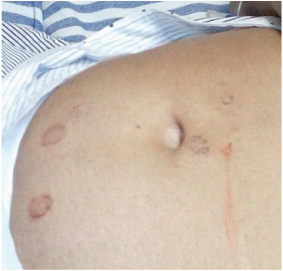
On examination, she was conscious and oriented; moderately built and nourished. Her heart rate was 130 beats/minute, blood pressure 140/90 mmHg and respiratory rate of 24 breaths/minute, with saturation 94% (in room air). She was febrile with temperature of 1030F. She was pale and icteric. There were purpuric spots over her body [Table/Fig-1]. Her respiratory system examination revealed minimal fine left basal crepitations. Other systemic examinations were normal.
Purpura spots over the abdomen.
Her complete blood count showed pancytopenia with hemoglobin 8.5g% (12-15), Packed Cell Volume (PCV) 25.6% (36–52), total counts 3300 cells/ cmm (4,000-10,000) with differential counts as Neuprophils 25% Lymphocytes 75%, platelets 54,000 cells/ cmm (150,000- 450,000), Mean Corpuscular Volume (MCV) 78.9fL (79-93), Mean Corpuscular Hemoglobin (MCH) 29.9pg/cell (26-32), MCHC 37.9g% (32-36) and Erythrocyte Sedimentation Rate (ESR) 15mm/ hour. Her blood sugars, electrolytes, calcium, thyroid profile, prothrombin time and activated partial thromboplastin time were normal. Her urea was 56mg% (20- 40) and creatinine 1.7 mg% (0.4-1.4). Her liver functions were deranged with total bilirubin 4.7mg% (0.3-1.3), direct bilirubin 2.0mg% (0.1– 0.4), Serum Glutamic Oxaloacetic Transaminase (SGOT) 456 IU/ L (12-38), Serum Glutamic Pyruvic Transaminase (SGPT) 147 IU/ L (7- 41), Alkakaline Phosphatase (ALP) 935 IU/ L (33-96), total protein 5.5gm% (6.7–8.6), albumin 3.5gm% (3.5–5.5) and Gamma Glutamyl Transferase (GGT) 40U/L (9-58). Her peripheral smear showed pancytopenia with no abnormal cells. Her urine microscopy showed only 3-4 pus cells, with no evidence of haematuria and urine culture showed scanty growth of E.coli. Her blood culture was sterile; Veneral Disease Research Laboratory test (VDRL) non-reactive; and Mantoux test, Human Immunodeficiency Virus (HIV), hepatitis A, hepatitis B, hepatitis C, malarial smear, dengue and leptospirosis serology, and Weil Felix were negative. Antinuclear antibody, Coombs’ test and serum protein electrophoresis were negative. Her stool occult blood was also negative. Electrocardiogram (ECG) showed sinus tachycardia and chest X-ray and Echocardiography (ECHO) heart was normal. Ultrasound abdomen showed mild hepatosplenomegaly. With the above mentioned information, a probable diagnosis of Systemic Inflammatory Response Syndrome (SIRS) with Multiorgan Dysfunction Syndrome (MODS) secondary to urinary tract infection, with underlying RA and ILD was considered. She was started on injection meropenum, which was also sensitive according to urine culture and sensitivity.
On day 2, she continued to be febrile continuously (with temperature of 1010F -1020F), despite being put on round the clock paracetamol. In view of a normal ESR in the midst of sepsis, the possibility of MAS was considered. Her serum ferritin was > 1650μg/L (15-200), fibrinogen 0.9 g/ L (1.5-3.0), triglycerides were 900mg/dl (< 150) and vitamin B12 levels were normal. Cerebrospinal fluid analysis showed total count of 12/cumm, all of which were lymphocytes, with normal protein and sugars. Bone marrow examination showed presence of haemophagocytes, thereby confirming the diagnosis of MAS (bone marrow sample was send to an outside laboratory for evaluation; and following the death of the patient, the relatives disposed off the slides and only the report was left). Epstein-Barr Virus (EBV) antibody test was negative. Intravenous immunoglobulin, dexamethasone and etoposide along with cotrimoxazole and fluconazole were started.
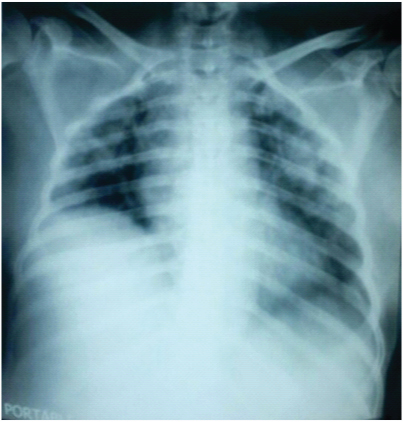
By day 3, her ESR further reduced to 7 mm/ hour; and pancytopenia, renal and liver parameters worsened. She had a drop in saturation and chest X-ray showed features of Acute Respiratory Distress Syndrome (ARDS) [Table/Fig-2], with hypoxia on Arterial blood gas (ABG) analysis. She was intubated and put on mechanical ventilation with ARDS protocol. On day 4, she continued to be febrile throughout, with blood pressure of 120/ 70 mmHg; and her blood counts showed Hb of 7.0g%, total counts 1200/cumm (N32 L68) and platelets 27,000/cumm. Urea and creatinine levels worsened to 106 mg/ dl and 3.1mg/ dl respectively. Liver functions remained almost the same. Injection filgrastim was added. On day 5 of admission, patient went into cardiac arrest and died.
Chest X-ray showing bilateral pulmonary infiltrates suggestive of ARDS.
Discussion
MAS is a life threatening complication of RA and other childhood systemic inflammatory disorders. It may be considered as a form of secondary or reactive HLH, where there is an uncontrolled proliferation and activation of T lymphocytes and well-differentiated macrophages, thereby resulting in haemophagocytosis and cytokine overproduction.
HLH has two forms: Primary or familial HLH, which is autosomal recessive; and secondary HLH seen in association with viral infections (especially EBV), malignancies and autoimmune diseases.
The diagnostic criteria outlined by the Histiocyte Society comprises of the following:
1) Fever,
2) Splenomegaly,
3) Cytopenias affecting at least two of three lineages in the peripheral blood,
4) Hypertriglyceridemia (≤ 265 mg/dl) and/or Hypofibrinogenaemia (≥ 1.5 g/L),
5) Haemophagocytosis in bone marrow, spleen, or lymph,
6) Low or absent NK-cell activity,
7) Hyperferritinaemia (>500 mg/L), and
8) High levels soluble CD25 (>2,400 U/ml).
The diagnosis requires five out of the eight criteria to be fulfilled; but patients with a molecular diagnosis consistent with HLH do not necessarily need to fulfil the diagnostic criteria. Genetic mutations involving PRF, SH2-DIA, UNC13D and STX11 have been linked to familial HLH. Cerebrospinal fluid analysis in HLH reveals pleocytosis and/or elevated spinal fluid protein, while liver biopsy resembles chronic persistent hepatitis [1–3]. The patient being described had fulfilled six of the above mentioned criteria; i.e., fever, spenomegaly, pancytopenia, hypertriglyceridemia and hypofibrinogenaemia, haemophagocytosis in bone marrow and hyperferritinaemia.
The treatment of HLH consists of different phases. During the induction phase etoposide and dexamethasone are used [4]. Salvage therapy using infliximab, daclizumab, alemtuzumab and anakinra have been tried when patients do not show at least a partial response within 2-3 weeks of induction therapy [5–8]. Prophylaxis against Pneumocystis jirovecii and fungal infections, intravenous immunoglobulin, and neutropenic precautions should also be considered. The continuation phase consists of pulses of dexamethasone and etoposide. Cyclosporine may be added in patients with stable vitals and normal liver and kidney function. Hematopoietic stem cell transplantation is recommended in the case of familial HLH, recurrent or progressive disease despite intensive therapy, and central nervous system involvement [4]. Antithymocyteglobulin with prednisolone have been tried in patients who have failed to respond to etoposide based regimens [6].
Conclusion
Systemic Inflammatory Response Syndrome (SIRS) is an important differential diagnosis for MAS. However, the presence of hyperferritinaemia and falling ESR should raise the suspicion of MAS. Though MAS is a rare complication of RA, this case highlights the importance of considering MAS in all cases of continuous fever with pancytopenia and low to normal ESR, especially in those with autoimmune disease. The occurrence of ARDS in MAS has rarely been reported.
[1]. Henter J-I, Elinder G, Ost A, The FHL study group of the histiocyte society. diagnostic guidelines for haemophagocytic lymphohistiocytosisSemin Oncol 1991 18:29-33. [Google Scholar]
[2]. Janka GE, Schenider EM, Modern management of children with haemophagocy ticlymphohistiocytosisBr J Haematol 2004 124:4-14. [Google Scholar]
[3]. Henter J-I, Tondini C, Pritchard J, Histiocytic syndromesCrit Rev Oncol Hematol 2004 50:157-74. [Google Scholar]
[4]. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL, How I treat haemophagocytic lymphohistiocytosisBlood 2011 118(15):4041-52. [Google Scholar]
[5]. Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F, Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroidsJ Clin Rheumatol 2011 17(1):23-27. [Google Scholar]
[6]. Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, Gondo H, Imashuku S, Success with infliximab in treating refractory haemophagocytic lymphohistiocytosisAm J Hematol 2006 81(1):59-61. [Google Scholar]
[7]. Olin RL, Nichols KE, Naghashpour M, Wasik M, Shelly B, Stadtmauer EA, Successful use of the anti-CD25 antibody daclizumab in an adult patient with haemophagocyticlymphohistiocytosisAm J Hematol 2008 83(9):747-49. [Google Scholar]
[8]. Strout MP, Seropian S, Berliner N, Alemtuzumab as a bridge to allogeneic SCT in atypical haemophagocytic lymphohistiocytosisNat Rev Clin Oncol 2010 7(7):415-20. [Google Scholar]