Chromosomal abnormality is a condition in an organism or a cell where the structure of any chromosome or the number of chromosomes differs from the normal karyotype. Cytogenetic analysis provides a genome-wide snapshot of an individual’s chromosomes by the process of pairing and arranging all of them in an order, and can reveal changes in chromosome numbers (aneuploids) and more delicate structural changes (chromosomal deletions, duplications, translocations and inversions) in the autosomes or sex chromosomes. In medical genetics, cytogenetic analysis is becoming an essential source of diagnostic information and evaluation of specific birth defects, genetic disorders, developmental delay, intellectual disabilities, and even cancers [1]. As yet, around 1000 chromosomal disorders have been reported [2]. Previous studies demonstrated that chromosomal abnormalities affected about 0.5% of the live births, contributing significantly to the birth defects and is a major cause of neonatal morbidity and mortality [3].
Chromosomal abnormalities are particularly common in cases of spontaneous abortions [4]. Of all the recognised conceptions at least 20% were estimated to be lost spontaneously, and half of them had chromosomal abnormality, mainly autosomal trisomy [5]. Cytogenetic analysis of gametes revealed that 10% of spermatozoa and 25% of mature oocytes were chromosomally abnormal. Between 1 to 3% of all the recognised conceptions are triploids. Children suspected for chromosomal disorders have shown wide range of chromosomal aberrations [6–8]. During the last decade, cytogenetic analysis has become a very important tool for genetic counselling, which deals with the human problems associated with the occurrence or risk of a genetic disorder in a family and helps to understand the diagnosis, prognosis and available management, the genetic basis and chance of recurrence and the options available. The availability of prenatal genetic testing is gaining popularity recently, and helping many couples at high genetic risk to embark upon pregnancies. But still some structural abnormalities cannot be detected before birth and remains as an important problem and requires early intervention. Though the prenatal diagnosis for chromosomal abnormalities have progressed rapidly, patients with chromosomal abnormalities remains as an important medical problem today [7].
Our study aimed to evaluate the cytogenetic findings in patients referred for suspected chromosomal abnormalities. Further, we studied the frequency of chromosomal abnormalities in these patients and compared our results with those reported elsewhere. This study can enhance awareness among paediatrician/gynaecologist for proper management and genetic counselling.
Materials and Methods
This retrospective study was carried out to identify the frequency and pattern of chromosomal aberrations among patients referred to the Division of Genetics, Department of Paediatrics, Maulana Azad Medical College and associated Lok Nayak Hospital, New Delhi over a period of 5 years (2010-2015). Before cytogenetic analysis a detailed interview was conducted to obtain the medical history of all the cases. Patients presented with multiple congenital anomalies, intellectual disabilities, mongoloid features, mental retardation and/or developmental delay, ambiguous genitalia, primary amenorrhea, blood disorders, short stature and parents of the individuals with structural chromosomal abnormalities were included in the study. A written informed consent was taken from children’s parent/guardian for cytogenetic testing. Based on the reason for referral, we categorized them into five groups: 1) Down’s syndrome (DS) group suspected for DS (Presented with specific clinical features like facial appearance, infants having brachycephaly, short neck, clinodactyly, single palmar creases, wide gap between the first and second toes, and hypotonia); 2) Turner syndrome (TS) group suspected for TS (Presented with consistent features like short stature and infertility from steak gonads, webbed neck, broad chest, cubitus valgus, coarctation of the aorta, renal anomalies and visual problems, associations with autoimmune thyroiditis, hypertension, obesity and non- insulin dependent diabetes in some cases); 3) Disorders of Sex Development (DSD) group (including genital ambiguity, disorders of gonadal (testicular) development, disorders of gonadal (ovarian) development, abnormal external genitalia in male, cryptorchidism, concealed penis, etc); and 4) Intellectual disability group and miscellaneous group (including developmental delay, Trisomy 18 (Edwards syndrome), intellectual disability, congenital heart diseases, etc)
For cytogenetic analysis, as per our hospital protocol, 5 ml peripheral blood sample was collected from all the patients and stored into heparinized test tubes. Chromosomal analysis was performed on cultured lymphocytes in culture medium in an incubator at 37°C for 72 hours. Metaphase harvesting was done by adding colcemid for 5 min following hypotonic KCl treatment for 1 hour and later fixation by using 3:1 methanol-acetic acid mix. Chromosomal analysis was performed according to the guidelines of the International System for Human Cytogenetic Nomenclature (ISCN, 2013) [9]. G-banding with Trypsin and Giemsa (GTG) (Seabright, 1971) [10] was used to determine the karyotypes, numerical as well as structural abnormalities were reported on at least 20 well-spread and well-banded metaphases after examination. Data was analysed using standard descriptive statistics. Patients identified with chromosomal abnormalities were given post test Genetic counselling.
Results
A total of 859 cases with suspected chromosomal abnormality were referred for cytogentic evaluation. Their ages ranged from newborn to 37 years with a mean age of 7.9±8.1 years, 256 (29.8%) cases were below 1 year age. Of the 859 cases, 491 (57.2%) were females and 368 (42.8%) were males. A total of 371 (43.1%) cases were found to be having chromosomal abnormalities. Numerical abnormalities were seen in 353 (41.0%) and structural abnormalities in 18 (2.0%) of the cases. Distribution of the cases based on clinical indications for referral of cytogenetic analysis was shown in [Table/Fig-1].
Distribution of the cases based on clinical indications for referral of cytogenetic analysis.
| Clinical indications | No of casesreferred n (%) | No of cases withChromosomalabnormalities n (%) |
|---|
| Down syndromegroup | 357 (41.6%) | 302 (81.4%) |
| Turner syndromegroup | 287 (33.4%) | 51 (13.7%) |
| Disorders of sexdevelopment group | 98 (11.4%) | 5 (1.3%) |
| Intellectual disabilitygroup and miscellaneousgroup | 117 (13.6%) | 13 (3.5%) |
| Total | 859 (100%) | 371 (100%) |
Based on the clinical features a total of 357 (41.6%) cases were suspected to be having DS, after successful karyotyping 302 (84.5%) were confirmed to be having DS out of which 176 (58.2%) were males and 126 (41.7%) females [Table/Fig-2]. The most common type of DS found was trisomy 21 due to non-disjunction in 258 (85.2%) cases, mosaicism in 32 (10.6%) cases followed by translocations in 12 (3.9%) cases. The common variety of translocation observed was between chromosomes 14 and 21 in 8 (66.7%) cases, followed by translocations between two chromosomes 21 in 4 (33.3%) cases. Various types of chromosomal abnormalities in DS Group were shown in [Table/Fig-3].
Showing the chromosomal complement of 47, XX, +21.
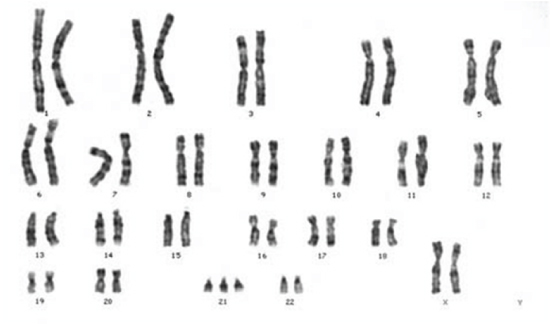
Chromosomal abnormalities in cases with down syndrome group.
| Karyotypes | No. of cases (%) |
|---|
| 47,XY,+21 | 150 (49.7) |
| 47,XX,+21 | 108 (35.8) |
| 46,XY/47,XY,+21 | 16 (5.2) |
| 46,XX/47,XX,+21 | 12 (4.0) |
| 46,XX/47,XY, +21 | 4 (1.3) |
| 46,XX,rob(14;21)(q10;q10),+21 or46,XY,rob(14;21)(q10;q10),+21 | 8 (2.7) |
| 46,XX,rob(21;21)(q10;q10),+21 or46,XY,rob(21;21)(q10;q10),+21 | 4 (1.3) |
Based on the clinical features a total of 287 (33.4%) cases were suspected to be having TS and 51 (17.7%) cases were confirmed to be having TS after successful karyotyping. Classical 45,X monosomy 13 (25.4%) were less than the Turners syndrome variants 38 (74.5%). TS variants includes 45,X/46,XX (70.5%) and 45,X/46,XY (3.9%). Chromosomal abnormalities in TS group were shown in [Table/Fig-4,5].
Chromosomal abnormalities in cases with Turner syndrome, Disorders of sex development, Intellectual disability group and miscellaneous groups.
| Karyotypes | No. of cases (%) |
|---|
| Turners syndrome (N=51) |
| 45,XO | 13 (25.4) |
| 45,XO/46,XX mosaic | 36 (70.5) |
| 45,XO/46,XY mosaic | 2 (3.9) |
| Disorders of sex development (N=98) |
| 46,XY | 66 (67.3) |
| 46,XX | 32 (32.7) |
| 45,X/46,XX | 1 (1.0) |
| 45,X/46,XY | 2 (2.0) |
| 46,XX/46,XY Chimerism | 2 (2.0) |
| Intellectual disability group and miscellaneous groups (N=13)Edwards syndrome |
| 47,XX,+18 Trisomy | 2 (15.3) |
| Klinefelter syndrome |
| 47,XXY | 4 (30.7) |
| 47,XYY | 1 (7.6) |
| Structural abnormalities |
| 46,XY,rob(13;14) (q10;q10) or46,XX,rob(13;14)(q10;q10) | 3 (23.0) |
| 45,XX,rob(13;14) (q12.1;q11.2) | 3 (23.0) |
Showing the chromosomal complement of 45, X.
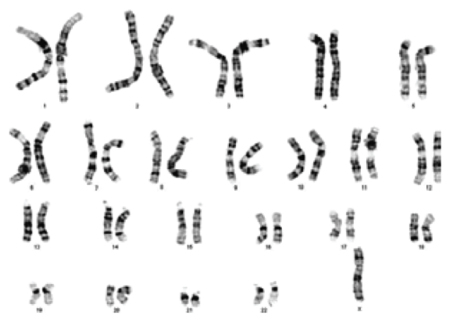
A total of 98 (11.4%) cases with Disorders of sex development were referred for karyotyping. 66 (67.3%) were successfully karyotyped and found to be male karyotype (46,XY) and 32 (32.7%) to be female karyotype (46,XX). The common chromosomal abnormalities observed in the cases with disorders of sex development were 45,X/46,XX 1 (1.0%), 45,X/46,XY 2 (2.0%), 46,XX/46,XY 2 (2.0%) and 47,XXY 1 (1.0%). Chromosomal abnormalities in disorders of sex development group were shown in [Table/Fig-4].
A total of 117 (13.6%) cases with intellectual disability and various congenital abnormalities, developmental delay, dysmorphic features and other miscellaneous cases were referred for karyotyping. A total of 13 (11.1%) were having abnormal karyotype among them 7 (53.9%) had numerical abnormalities i.e., 2 (15.3%) Edwards syndrome (47,XX +18 Trisomy), 4 (30.7%) Klinefelter syndrome (47,XXY), 1 (7.6%) 47,XYY [Table/Fig-6] and 6 (46.1%) had structural abnormalities i.e., 3 (23.0%) 46,XY,rob(13;14) (q10;q10) or 46,XX,rob(13;14)(q10;q10), 3 (23.0%) 45,XX,rob(13;14) (q12.1;q11.2). Chromosomal abnormalities in Intellectual disability and miscellaneous group were shown in [Table/Fig-4].
Showing the chromosomal complement of 47, XYY.
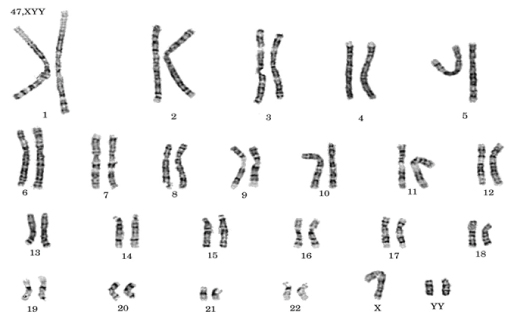
Discussion
In this present study, a total of 859 cases were evaluated for cytogenetic analysis of suspected chromosomal abnormalities who were referred to Division of Genetics, Department of Paediatrics. The prevalence of chromosomal abnormalities was found to be 42.4% in all these cases. Numerical abnormalities 41.0% were the majority of chromosomal abnormalities observed followed by structural abnormalities 2.0%. In our study, both children and adults were included in corroboration with previous studies who have reported the prevalence, and chromosomal abnormalities in the cases suspected with genetic disorders [4,7–11].
The reason for high prevalence of chromosomal abnormalities might be because of the legal prohibition on termination of pregnancies, even though advanced prenatal diagnostics techniques were available in our country. Cytogenetic analysis is an essential tool in genetic counselling to establish a definitive diagnosis, and to estimate the risk of recurrence of the chromosomal disorders in future pregnancies and deciding clinical management. It is gold standard, but definite diagnosis is achieved by MOL bio tests, for e.g., in case of deletions.
Down syndrome group
DS, due to trisomy 21, is the most common autosomal aneuploidy and was confirmed by karyotyping in our study [Table/Fig-3]. Our results were in consistent with many previous studies carried out in India by many investigators [Table/Fig-7] [2,12–21]. A cytogenetic analysis carried out in Hyderabad by Issac et al., in 1985 revealed DS in 96.5% affected cases and 3.5% had chromosomal mosaicism, they have shown the incidence of 1.17 per 1000 or 1 in 853 live births [22]. Patel and Adhia in 2005 studied the frequency of congenital malformations in 17,653 consecutive newborns in Mumbai, shown the incidence of DS was 1 in 1200 [23]. A study of malformations and DS in Baroda region on 31,775 births showed overall prevalence of 1.04 per 1000 and also increased DS prevalence was reported in maternal age (0.54 per 1000 at 15-19 years to 15.6 per 1000 at > 40 years) [24]. Similar results were shown by Balkan et al., in 2010 on a cytogenetic analysis of 4216 cases in South east Turkey showing highest frequencies of DS (84.8%) [21]. Male predominance was observed in the DS cases {176 (58.2%) males versus 126 (41.7%) females}. Similarly Kovaleva et al., in 2002 has done a meta-analysis from 55 publications showing the sex ratio between male and female cases in DS reported male predominance of trisomy 21 [25]. The probable genetic mechanism for male predominance include the joint segregation of chromosome 21 and Y chromosome in spermatogenesis and non-disjunction of chromosome 21 during second meiotic division in oogenesis caused by Y chromosome bearing spermatozoa [25]. Chromosomal non-disjunction is the main cause of DS with 85.2% of the cases having free trisomy 21, due to mosaicism 10.6% and translocations 3.9%. Similar findings were reported by Jyothy et al., in 2000 on a cytogenetic study of 1001 DS cases in Department of Human Cytogenetic, Hyderabad showing the frequency of free trisomy 87.9%, mosaicism 7.7% and translocations 4.4% [26]. Another study by Kaur et al., in 2010 on chromosomal abnormalities in 1950 cases referred with genetic disorders to Centre for Genetic Disorders, Amritsar reported the frequency of free trisomy, mosaicism and translocations as 90.5%, 3.1% and 2.7% respectively. In contrast, study conducted by Ahmed et al., in 2005 at Department of Paediatrics and Internal Medicine of Military Hospital, Pakistan [27] and Verma et al., in 1991 at Department of Paediatrics, All India Institute of Medical Sciences, New Delhi [28] reported higher frequency of translocation DS than the mosaic. Among various studies interestingly a variable pattern of DS is reported [5,13–16]. The variation may be because of selected study populations. Translocation DS cases usually indicates karyotypic analysis for both parents as either of them may be carriers for balanced translocation involving with chromosome 21. There is an increased risk of aneuploid offspring in every conception with translocation carriers and recurrence risk depends on the chromosomes that are fused and the sex of the carrier parents [29]. It has been reported from previous studies that maternal age is directly related to risk of trisomy 21, i.e., 1/1300 for a 25-year-old, 1/365 for a 35-year-old and the risk directly increases to 1/30 at the age of 45-year-old [29]. The recurrence risk is 100% if any one of the parents is the carrier of a balanced translocation involving the two 21 chromosomes. DS can lead to a myriad of symptoms related to physical, physiological and psychological impairments associated with many body systems. Acute lymphoblastic leukaemia, duodenal stenosis and Alzheimer’s are found to higher in DS than the general population [30]. In addition to indicating the recurrence risk, karyotyping is useful tool in the clinical follow-up of certain disorders associated with DS. By identifying DS karyotype, patient’s family can be given genetic counselling about susceptibility to acute leukaemia, duodenal stenosis, Alzheimer’s and associated symptoms. So that an early medical management can be initiated and increase patient’s life expectancy.
Comparison of the frequency of Chromosomal Aberrations of the Indian population of the present study with other Studies [2,12–21].
| Other Studies | Total number ofpatients examined | Frequency ofChromosomalAberrations found (%) |
|---|
| Singh et al., [12] (1977) | 451 | 130 (28.8%) |
| Winter et al., [13] (1980) | 140 | 60 (43.0%) |
| Verma and Dosik [14] (1980) | 357 | 97 (27.2%) |
| Mitra et al., [15] (1988) | 325 | 77 (23.6%) |
| Navasaria et al., [16] (1993) | 1000 | 166 (16.6%) |
| Hussain and Zaki [17] (1999) | 1000 | 134 (13.4%) |
| Hatem and Jamil [18] (2002) | 747 | 152 (20.3%) |
| Duarte et al., [19] (2004) | 916 | 269 (29.3%) |
| Goud et al., [20] (2005) | 1800 | 510 (28.3%) |
| Balkan et al., [21] (2010) | 4216 | 1360 (32.2%) |
| Thillainathan S et al., [2] (2015) | 1548 | 783 (50.6%) |
| Present study (2016) | 859 | 371 (43.1%) |
Turner syndrome group
In India the given birth rate is 22,000 and considering the incidence of TS to be 1 in 2,500 female births, each year ~ 5,200 girls with TS were born in India [31]. TS is the most common sex chromosomal aneuploidy accounting to 33.4% of cases and 13.9% of them were karyotypically confirmed TS in our study [Table/Fig-4]. Usually TS is diagnosed at the time of puberty because of the failure of sexual maturation resulting from ovarian dysgenesis. TS results from partial or complete absence of one of the two X chromosomes normally present in females. It may also result from deletion or duplication in the genetic material of X chromosome. TS variants (70.5%) were found to be higher than the classical 45, X monosomy (25.5%) in our study. Similarly Rajasekhar et al., in 2010 reported higher proportion of mosaics TS than the classic TS [32]. Duarte et al., in 2004 examined the chromosomal composition of 916 patients in Brazil and reported most common TS mosaic (53.6%) than classical monosomy 45, X (28.6%) [19]. In contrast Suri et al., in 1995 showed that the commonly observed karyotype was classical 45, X monosomy (44.4%) than the TS mosaic (24.4%) [33]. The reason for these variations may be because of the referral reasons of the study populations.
Disorders of sex development group
The condition of imperfect sexual differentiation into either male or female is called intersexuality. Sexual ambiguity is due to several disorders of gonadal differentiation, male and female pseudo her-maphroditism and hermaphroditism. Approximately 1 in 4500 live born infants has a severe genetic anomaly to differentiate the sex and needs immediate assignment of sex [34]. 9.1% cases had discrepancy between the phenotypic sex and genetic sex in our study [Table/Fig-4]. They had normal female phenotype with 46, XY chromosome suggesting sex reversal condition. Rajasekhar et al., in 2010 reported similar results showing the frequency of 14.9% of XY females [32], whereas Sangeetha et al., in a study on sexual ambiguity in Humans reported higher frequency of 33.3% of XY females [34]. The reason for higher frequency may be because inclusion of both children and adults in the study.
Intellectual disability group and miscellaneous group
Mental retardation is the incomplete development of mental capacities and associated behavioural abnormalities [35]. Chromosomal abnormalities are the main cause of mental retardation. Every year nearly 7.6 million children were born globally with severe congenital malformations and 90% of mentally retarded people were born in low income countries [36]. A total of 117 cases with various congenital abnormalities, dysmorphic features, developmental delay and intellectual disabilities were studied, in which 13 (11.1%) had different chromosomal abnormalities detected by cytogenetic analysis [Table/Fig-4]. Edward syndrome (Trisomy 18) occurs due to non-disjunction and the incidence increases with maternal age. The overall incidence of trisomy 18 is around 1 in 6000 live births, majority of conceptions are lost spontaneously with only about 2.5% surviving to term [37]. In this study, only 2 (15.3%) of Edward syndrome was reported and no cases of Patau syndrome were identified. Klinefelter syndrome arises by non-disjunction and the additional X chromosome is equally likely to be maternally or paternally derived. The incidence of Klinefelter syndrome is 1 in 600 live born males. There is no increased early pregnancy loss associated with this karyotype [37]. A 30.7% were diagnosed with 47, XXY karyotype in our study with the primary features of this syndrome and 7.6% were with 47, XYY syndrome. A 46.1% had Robertsonian translocations detected by conventional GTG-banded cytogenetic analysis in this group.
Limitation
Our study has certain limitations; G-banded chromosome analysis could not identify single gene defects or complex structural rearrangements in normal chromosomal compliment.
Conclusion
In conclusion, the genetic diseases/ chromosomal disorders can be controlled by integrated and comprehensive efforts with strategies including best possible treatment and prevention by community education, genetic counselling, population screening and an early diagnosis of genetic disorders. The genetic counselling helps families to cope up with emotional, psychological and medical consequences of genetic disorders. There are insufficient data in current epidemiology of genetic disorders in India. This data can generate information on the importance of genetic counselling and the demand for genetic services in India. The efficient genetic registries, genetic databases and continuous investments in genetic research are key to successful public health interventions.