Risk Stratification in Paragangliomas with PASS (Pheochromocytoma of the Adrenal Gland Scaled Score) and Immunohistochemical Markers
Maithili Mandar Kulkarni1, Siddhi Gaurish Sinai Khandeparkar2, Sanjay D. Deshmukh3, R.R. Karekar4, Vandana L. Gaopande5, Avinash R. Joshi6, Mrunal V. Kesari7, R.R. Shelke8
1 Associate Professor, Department of Pathology, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
2 Associate Professor, Department of Pathology, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
3 Professor, Department of Pathology, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
4 Associate professor, Department of Medicine, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
5 Professor, Department of Pathology, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
6 Professor, Department of Pathology, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
7 Assistant Professor, Department of Pathology, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
8 Professor, Department of Surgery, Shrimati Kashibai Navale Medical College and General Hospital, Pune, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Maithili Mandar Kulkarni, E 401, Samrajya, Shivtirthnagar, Paud Road, Kothrud, Pune, Maharashtra, India.
E-mail: drmaithili@rediffmail.com
Introduction
Paragangliomas (PGLs) are rare tumours that arise in sympathetic and parasympathetic paraganglia and are derived from neural crest cells. Presence of metastasis is the only absolute criterion for malignancy. There is no single histo-morphological feature indicating malignant potential and multiple parameters have been proposed to prognosticate the individual case. This includes studies conducted using Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) and Immunohistochemical (IHC) markers.
Aim
We have studied ten cases of paraganglioma and attempted to correlate the prognosis with multiple clinicopathological variables.
Materials and Methods
This study was done in a tertiary care general hospital over a period of five years. Available clinical records and histopathology slides of all patients were reviewed. Using Pheochromocytoma of the Adrenal Gland Scaled Score (PASS), we divided the cases into two groups-tumours showing high risk behaviour (PASS≥4) and tumours showing benign behaviour (PASS<4). IHC analysis was done using synaptophysin, chromogranin, S100 and Ki67. We correlated S100 immunoreactivity and Ki67 proliferative index with PASS score. Both PASS score and IHC markers were also correlated with clinical outcome.
Results
There were six Pheochromocytomas (PHC) and four Paragangliomas (PGL). Two paragangliomas were retroperitoneal and one each was located in ear (HNPGL) and broad ligament. PASS score was ≥4 in five cases and <4 in five cases. Out of five cases in which PASS was ≥4, three cases showed clinical evidence of malignancy and two cases were benign. All the cases in which PASS was <4 were clinically benign. S100 immunoreactivity was grade 1 in two cases, grade 2 in six cases and grade 3 in two cases. The cases in which S100 immunoreactivity was grade 1 were malignant. One case in which S100 was grade 2 was clinically malignant. Ki67 labeling index was raised (>3%) in two cases, which were malignant correlated with malignant PASS score.
Conclusion
We conclude that the following clinicopathological parameters should be taken into account for risk assessment of malignant behaviour of paragangliomas- location, size, PASS score, S100 immunoreactivity and Ki67 labeling index.
Paraganglioma, PASS score pheochromocytoma, S100 immunoreactivity
Introduction
Paragangliomas (PGLs) are rare neuroendocrine tumours that arise in sympathetic and parasympathetic paraganglia and are derived from neural crest [1]. 80 to 85% of these tumours arising from adrenal medulla are known as pheochromocytomas (PHCs) The remaining 15 to 20% arising from the extra adrenal chromaffin tissue are referred to as secreting paragangliomas (sPGLs). Paragangliomas derived from parasympathetic tissue in the head and neck one labeled as head and neck paragangliomas (HNPGLs) [2]. PHCs and abdominal sPGLs are usually catecholamine producing tumours, whereas most of the HNPGLs are non-functioning [1]. The term pheochromocytoma was coined by Poll in 1905 to describe the dusky (pheo) colour (chromo) cut surface of the tumour when exposed to dichromate [3]. The only absolute criterion of malignancy is evidence of metastatic tumour spread [4]. Malignant PGLs are extremely rare and the estimated incidence in USA in 2002 was 93 cases per 400 million persons [5]. Very few studies evaluating the clinical presentation of PHCs are available in Indian literature. In one study, out of 10 cases of PGLs found over a period of 10 years, only 2 showed evidence of malignancy [6]. About 10% and 20% of PHCs and abdominal sPGLs respectively, are malignant [7] while, HNPGLs are usually benign [8].
There is no single histomorphological feature indicating the malignant potential and a number of multifactorial systems have been proposed. The cytomorphometric study done by some researchers showed that extra-adrenal location, coarse nodularity, confluent necrosis and absence of hyaline globules were associated with malignant behaviour [9]. Another study highlighted the significance of tumour size and necrosis, and not vascular or local invasion [10]. Several scoring systems considering invasion, histologic growth patterns, mitotic activity have been used to stratify the risk [11–13]. One of the most utilized score is Pheochromocytoma of Adrenal Gland Scales Score (PASS) proposed by Thompson in 2002 [14]. [Table/Fig-1] shows the parameters and their values which are necessary to calculate the PASS. A PASS score <4 or ≥ 4 suggest benign versus malignant lesion respectively. We have studied a series of paraganglioma to correlate the biological behaviour with multiple clinicopathological parameters.
Pheochromocytoma of the adrenal gland scoring scale (PASS) [11].
| Histomorphological parameter | Score |
|---|
| Nuclear hyperchromasia | 1 |
| Profound nuclear pleomorphism | 1 |
| Capsular invasion | 1 |
| Vascular invasion | 1 |
| Extension into periadrenal adipose tissue | 2 |
| Atypical mitotic figures | 2 |
| Greater than 3 mitotic figures/10 high-power field | 2 |
| Tumour cell spindling | 2 |
| Cellular monotony | 2 |
| High cellularity | 2 |
| Central or confluent tumour necrosis | 2 |
| Large nests or diffuse growth (>10% of tumour volume) | 2 |
| Total | 20 |
Materials and Methods
This study was conducted in a tertiary care hospital for a period of five years (2010 to 2014). Records of ten patients diagnosed as PGLs were retrieved from the archives. Histopathological data and intraoperative findings were mainly studied. Follow up information was obtained by written questionnaires and direct communication with the treating physicians or the patients. Haematoxylin and eosin stained slides from all patients were reviewed by two pathologists for morphologic assessment. PASS score was assigned to each case dividing tumours into two groups PASS ≥ 4 and PASS < 4. IHC was done on sections from tissue obtained by tissue microarray technique (TMA) [15]. Antigen retrieval was done in a pressure cooker in citrate buffer. IHC profile for synaptophysin (Sy38, Dako, cytoplasmic), chromogranin (Dako, cytoplasmic), S100 (Dako, cytoplasmic and nuclear), and Ki67 (MM1, Novacastra, nuclear) was studied in all cases. Pretreatment with citrate buffer was done. Primary antibodies were ready to use with optimum dilution and incubation period was 30 minutes. Immunoreactivity was graded as weak (1+), moderate (2+) and strong (3+) for S100 immunomarker. The reactivity was based on the number of sustentacular cells postitive for S100. The mean count was calculated from 10 high power fields (HPF). Ki67 labeling index immunoreactivity was calculated as the percentage of tumour cells positive for the proliferation marker Ki67. The proliferative index was expressed as a percentage of positively stained cells. Ki67 labeling index of 3 % was taken as cutoff to differentiate between benign and malignant tumours [16]. Tumours were classified into benign and malignant category based on the clinical findings and presence of metastasis. Comparison of multiple clinicopathological parameters with clinical outcome was done [Table/Fig-2].
Comparison of multiple clinicopathological parameters with clinical outcome.
| Case no | Site | Size(cm) | PASSScore | S100 | Ki67 | Characteristic feature | Clinical outcome |
|---|
| 1 | Adrenal | 6x4x4 | 6 | Gr2 | 1% | HG | B |
| 2 | Adrenal | 7x6x4 | 3 | Gr2 | 0% | HG | B |
| 3 | Adrenal | 6x4x3 | 6 | Gr2 | 0% | HG | B |
| 4 | Ear | 2x2x1 | 2 | Gr2 | 0% | - | B |
| 5 | Adrenal | 3x3x1 | 2 | Gr3 | 1% | HG | B |
| 6 | RP | 15x10x6 | 6 | Gr1 | 8% | Size >10cm | M |
| 7 | Adrenal | 4x5x4 | 6 | Gr1 | 1% | Capsular invasion | M |
| 8 | RP | 5x4x3 | 8 | Gr2 | 5% | Focal confluent necrosis | M |
| 9 | Broad ligament | 6x4x2 | 3 | Gr3 | 0% | Cystic degeneration | B |
| 10 | Adrenal | 4x3x2 | 2 | Gr2 | 0% | Cystic degeneration | B |
Results
There were six females and four male patients whose age ranged between 12 to 65 years and comprised of nine adults and one adolescent. Overall, there were six cases of PHCs and four cases of PGLs. The two PGLs were retroperitoneal and one each was located in ear (HNPGL) and broad ligament. Palpitation was the most common presentation. Hypertension was present in all cases except in two cases, one each of HNPGL and broad ligament PGL. Other clinical features seen were chest pain, breathlessness and pain in the abdomen. Laboratory values were available in eight patients who exhibited elevated levels of catecholamines, norepinephrine and epinephrine.
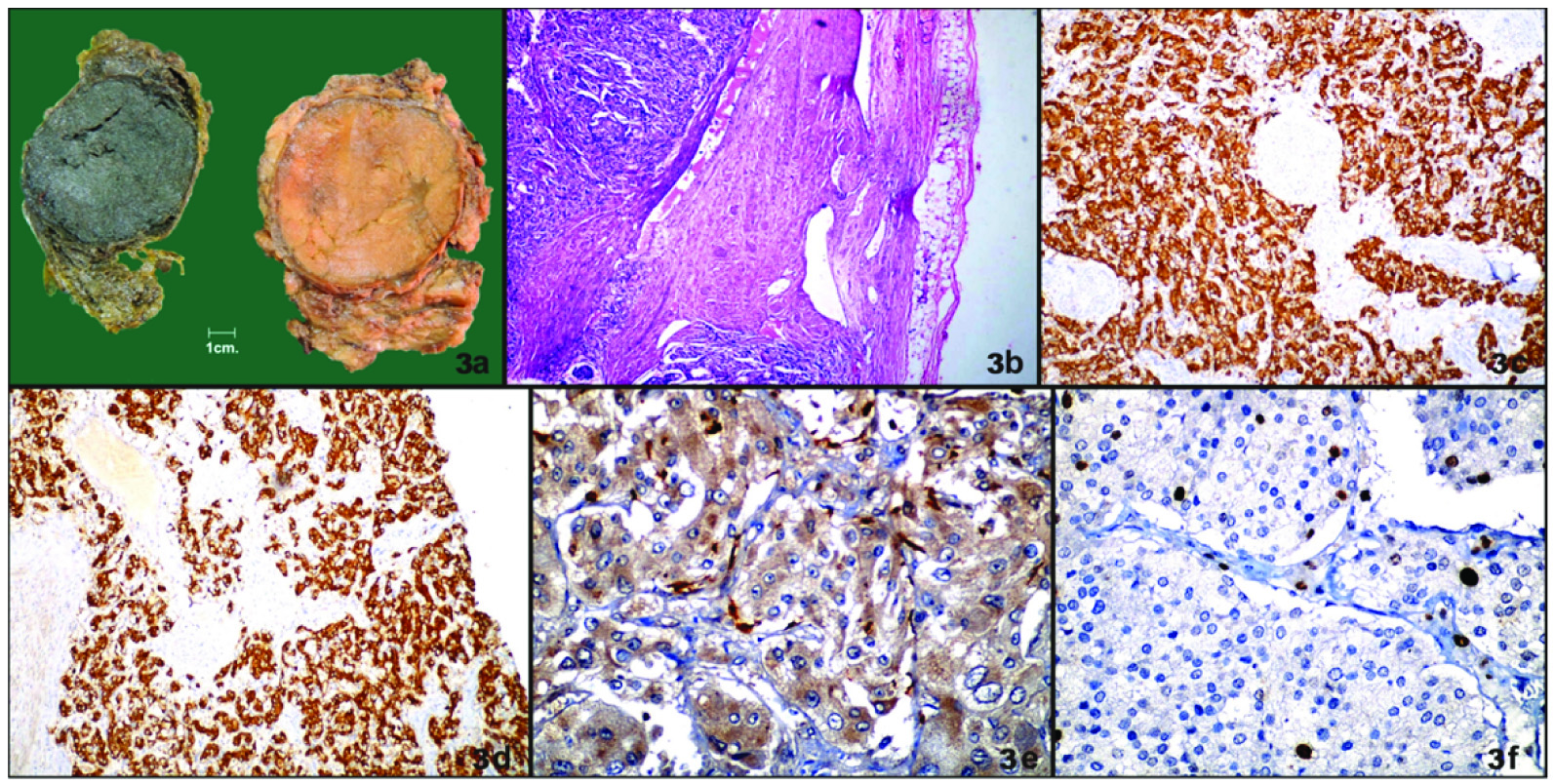
All tumours were encapsulated and well-circumscribed masses with positive dichromate reaction [Table/Fig-3a]. Two tumours showed variegated cut surface showing areas of haemorrhage or degenerative change. They were soft, grayish to brown red. The largest tumour size was 15x10x6 cm which was present in retroperitoneum. The smallest tumour size was 1x1cm present in ear. The tumour in broad ligament showed extensive cystic degeneration.
a-Gross photograph of pheochromocytoma with dichromate reaction. Photomicrographs of pheochromocytoma showing: b - capsular invasion (H and e; x100), c - chromogranin immunoreactivity (x100), d- synaptophysin immunoreactivity x(100), e- grade 2 S100 immunoreactivity (x400), f- Ki67 labeling index (8%) (x400).
RP = Retroperitoneal; Gr = grade; B = Benign; M = malignant; HG =Hyaline globules.
Histopathology
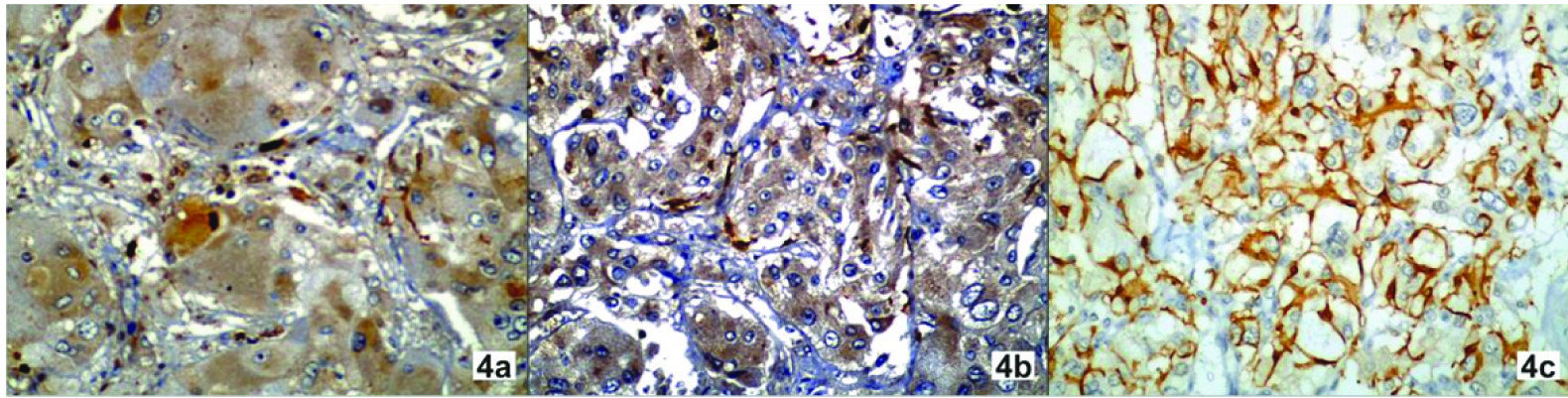
Majority of the tumours were well circumscribed and well encapsulated. The tumour cells were arranged in characteristic zell-ballen architecture. Intracytoplasmic hyaline globules were seen in four cases which were clinically benign. Degenerative changes like haemorrhage were seen. Vascular invasion was absent in all cases. Capsular invasion was noted in one case that had aggressive behaviour [Table/Fig-3b]. One case showed focal confluent necrosis and aggressive behaviour clinically. PASS score was ≥4 in five cases and <4 in five cases. Out of five cases in which PASS was ≥4, three cases were clinically malignant and two cases were clinically benign. All the cases in which PASS was <4 were clinically benign. All cases showed immunoreactivity for chromogranin [Table/Fig-3c] and synaptophysin [Table/Fig-3d]. Ki67 labeling index was raised (>3%) in two cases [Table/Fig-3f] and both cases were clinically malignant. It was <3% in eight cases, one of which was malignant. S100 staining revealed the presence of elongated cells. S100 immunoreactivity was grade 1 in two cases, grade 2 in six cases and grade 3 in two cases [Table/Fig-4]. Two cases in which it was decreased had evidence of malignancy and one case in which it was moderate was malignant. Cases were followed up for three years. Complete reversal of clinical (hypertension) and biochemical parameters (catecholamine levels) was noted in all benign and two malignant cases and partial reversal was noted in one case which on follow up showed disseminated disease. Out of three malignant cases one presented with disseminated disease and succumbed to death as a result of multiple metastases after one year.
Phtotomicrograph of pheochromocytoma showing S100 immunoreactivity: a=grade 1, b= grade 2, c= grade 3 (x400).
Discussion
Pheochromocytomas are uncommon tumours of sympathochromaffin system. The histologic diagnosis is relatively easy due to the characteristic morphological pattern. However, distinction of a benign from a malignant neoplasm has not been easy based on histological criteria alone. There is no gender predilection for pheochromocytoma. But, in this study male to female ratio was found to be 2:3. In this study, the mean age was 37 years while one case occurred in adolescent age group. Signs and symptoms for PGLs are non-specific, however hypertension was the most consistent sign observed in our study, except in one case of HNPGL [14]. There was equal distribution on right and left sides.
PASS score of <4 had benign behaviour and all malignant cases had PASS ≥ 6. Surprisingly, two cases in which PASS was 6 had benign behaviour. Thus PASS score between 4 and 6 need long term follow up. Recent studies suggest the values to be <4 for benign tumours and ≥6 for malignant tumours, whereas a value between 4 and 6 suggests an intermediate risk [17]. In one study done on 100 cases it was noticed that PASS can be used to separate tumours with a potential for a biologically aggressive behaviour (PASS≥4) from those that behave in a benign fashion (PASS <4) [14].
In a study done on 11 patients, it was found that PASS score ≥ 4 identifies malignant pheochromocytoma with a sensitivity 50% and specificity 45%. The authors suggested that PASS helps to reserve the more aggressive treatment and narrow the follow up for potentially malignant tumour [18]. Size and weight of the PHCs are directly related to PASS and malignancy [19].
PHCs are made up of dual cell population. Chief cells constitute majority of the tumour and are easily detected in H and E stained section. However, sustentacular cells which are less in number mandate special stains for accurate identification.
Few studies on S100 expression in paragangliomas are available in the literature. In present study, S100 protein immunoreactivity was observed in both the nucleus and cytoplasm of sustentacular cells surrounding the chromaffin cells [20,21]. In one study, heavy intense staining of the sustentacular cells was present only to become weak or absent in areas of large nest formation or diffuse growth pattern [14]. There has been a remarkable decrease in the immunoreactivity of S 100 protein in malignant cases [16,17,19–21,22]. It has been documented in some studies, that the absence of sustentacular cells in PHC is indicative of a greater potential for metastasis [20,21]. Sustentacular cells, when they are present in a malignant PGL, are an integral part of the tumour [21]. S100 positive cells were noted in metastatic deposits, suggesting that the sustentacular cells had metastatic potential along with chromaffin cells and are an integral part of the tumour [14]. A number of studies have addressed the issue of correlation of proliferation index with malignant potential based on the calculation of a Ki67 index using the antibody MIB1. A high proliferative labeling index of Ki67 >3% is considered a useful parameter for predicting malignant potential [23]. In our study two cases showed low sustentacular cell count. One case was of malignant retroperitoneal paraganglioma (size more than 10 cm, PASS score six, Ki67 index 8%). Other case was malignant PHC in which PASS score was 6, Ki 67 index was 1%. The third malignant case in which there was capsular invasion was extraadrenal paraganglioma (PASS was 6; sustentacular cell count was normal, Ki67 index was 8%).
Our observations indicate that combination of PASS score, S100 and Ki 67 labeling index is a better approach to prognosticate the case and should be the routine protocol in each case encountered. Similar observations are proposed in a series of 53 cases published by de wailly P et al. They suggested that presence of tumour necrosis, Ki-67 index >4% and S100 absence impose a close histopathological evaluation and follow up with regard to cases presenting a high risk of malignancy/recurrence [19].
Limitation
Ours is a study of small number of cases; therefore cumulative data added by other workers especially on S 100 expression which could be used as a potential marker of malignant transformation will pave the way for refined approach to risk stratification in cases of paraganglioma.
Conclusion
Malignant PGLs, as defined by the presence of metastasis, are very rare and aggressive tumours. As metastasis can develop several years after surgical removal of primary tumour, studies comparing benign and malignant PGLs need a long clinical follow up of patients. Findings in PASS scoring and IHC study with S100 and Ki67 labeling index can be used together for prognostication of paraganglioma.
[1]. Lenders JW, Eisenhofer G, Mannelli M, Pacak K, PhaeochromocytomaLancet 2005 366:665-75. [Google Scholar]
[2]. Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giachè V, Mannelli M, Updated and New Perspectives on Diagnosis, Prognosis, and Therapy of Malignant Pheochromocytoma/ParagangliomaJournal of Oncology 2012 2012:872713 [Google Scholar]
[3]. Samaan NA, Hickey RC, Shutts PE, Diagnosis, localization and management of pheochromocytoma - pitfalls and follow-up in 41 patientsCancer 1988 62:2451-60. [Google Scholar]
[4]. McNicol AM, Histopathology and immunohistochemistry of adrenal medullary tumours and paragangliomasEndocrine Pathology 2006 17:329-36. [Google Scholar]
[5]. Welander J, Söderkvist P, Gimm O, Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomasEndocr Relat Cancer 2011 18:R253-76. [Google Scholar]
[6]. Krishnappa R, Chikaraddi S B, Arun H N, Deshmane V, Pheochromocytoma in Indian patients: A retrospective studyIndian J Cancer 2012 49:188-93. [Google Scholar]
[7]. Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB, The diagnosis and management of malignant phaeochromocytoma and paragangliomaEndocr Relat Cancer 2007 14:569-85. [Google Scholar]
[8]. Lee JH, Barich F, Karnell LH, Robinson RA, Zhen WK, Gantz BJ, American College of Surgeons Commission on Cancer; American Cancer Society. National Cancer Data Base report on malignant paragangliomas of the head and neckCancer 2002 94:730-37. [Google Scholar]
[9]. Lewis PD, A cytophotometric study of benign and malignant phaeochromocytomasVirchows Arch B Cell Pathol 1971 9:371-76. [Google Scholar]
[10]. Nativ O, Grant CS, Sheps SG, Otallon J, Farrow G, van Heerden A, The clinical significance of nuclear DNA ploidy pattern in 184 patients with pheochromocytomaCancer 1992 69:2683-87. [Google Scholar]
[11]. Sherwin RP, Histopathology of pheochromocytomaCancer 1959 12:861-77. [Google Scholar]
[12]. Sisson JC, Shapiro B, Beierwaltes WH, Glowniak JV, Nakajo M, Mangner TJ, Radiopharmaceutical treatment of malignant pheochromocytomaJ Nucl Med 1984 25:197-206. [Google Scholar]
[13]. Symington T, Goodall AL, Studies in pheochromocytoma: I. pathologic aspectsGlas Med J 1953 34:75-96. [Google Scholar]
[14]. Thompson L, Pheochromocytoma of the of the Adrenal Gland Scaled Score (PASS) to separate benign from malignant neoplasms:a clinicopathologic and immunophenotypic study of 100 casesAm J Sur Pathol 2002 26:551-66. [Google Scholar]
[15]. Singh DK, Sakhuja P, Gondal R, Making and using inexpensive manually constructed tissue micro-array: Experience of a tertiary care hospital in IndiaIndian J Pathol Microbiol 2009 52:304-09. [Google Scholar]
[16]. Liu TH, Chen YJ, Wu SF, Gao J, Jiang WJ, Lu ZH, [Distinction between benign and malignant pheochromocytomas]Zhonghua Bing Li Xue Za Zhi 2004 33:198-202. [Google Scholar]
[17]. Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, “Prognostic indicators of malignancy in adrenal pheochromocytomas:clinical, histopathologic, and cell cycle/apoptosis gene expression analysisSurgery 2008 143:759-68. [Google Scholar]
[18]. Milka M, Kourda N, Zorgati MM, Bahri S, Ben Ammar S, Zermani R, Prognostic value of Pheochromocytoma of the Adrenal Gland Scaled Score (Pass score) tests to separate benign from malignant neoplasmaTunis Med 2013 91:209-15. [Google Scholar]
[19]. de wailly P, Oragano L, Rade F, Beaulieu A, Arnault V, Levillain P, Malignant pheochromocytoma: new malignant criteriaLangenbecks Arch Surg 2012 397:239-46. [Google Scholar]
[20]. Lloyd RV, Blaivas M, Wilson BS, Distribution of chromogranin and S100 protein in normal and abnormal adrenal medullary tissuesArch Pathol Lab Med 1985 109:633-35. [Google Scholar]
[21]. Unger P, Hoffman K, Pertsemlidis D, Thung S, Wolfe D, Kaneko M, S100 protein-positive sustentacular cells in malignant and locally aggressive adrenal pheochromocytomasArch Pathol Lab Med 1991 115:484-87. [Google Scholar]
[22]. Schlumberger M, Gicquel C, Lumbroso J, Tenenbaum F, Comoy E, Bosq J, Malignant pheochromocytoma: clinical, biological, histologic and therapeutic data in a series of 20 patients with distant metastasesJ Endocrinol in Invest 1992 15:631-42. [Google Scholar]
[23]. Tato A, Orte L, Diz P, Quereda C, Ortuno J, Malignant pheochromocytoma, still a therapeutic challengeAm J Hypertens 1997 10:479-81. [Google Scholar]