The spindle cell variant of embryonal rhabdomyosarcoma is a rare and a better differentiated variant of embryonal rhabdomyosarcoma, having a better prognosis compared to other types of rhabdomyosarcomas. So, it needs to be distinguished from classical forms of the neoplasm. Its morphological resemblance to spindle cell neoplasms like leiomyosarcomas and fibrosarcomas may pose diagnostic difficulties for the pathologist. This problem can be overcome by careful search for rhabdomyoblasts in sections, which are usually few, and Immunohistochemistry for myogenin. In the present case, a 15-year-old female presented with a progressively increasing swelling in the right upper eyelid, which was diagnosed as a rare variant of rhabdomyosarcoma. We have also attempted to discuss its differential diagnosis, and to emphasize the fact that this rare entity may be misdiagnosed.
Prognosis, Leiomyosarcoma, Orbit
Case Report
A 15-year-old female presented to the ophthalmologist with a progressively increasing swelling in the right upper eyelid since 2 months. The swelling measured 3x2 cm, it was painless and the patient’s vision was unaffected. An examination of the eye did not reveal any abnormality. A CT scan was advised and it showed a well defined enhancing extraconal mass measuring 2x2 cm between the medial orbital wall and the lateral wall of the globe. The adjacent bone was normal. The possibility of the lesion being a vascular neoplasm or a lymphoma was suggested by the radiologist. The mass was excised under general anaesthesia and the specimen was sent for histopathological examination.
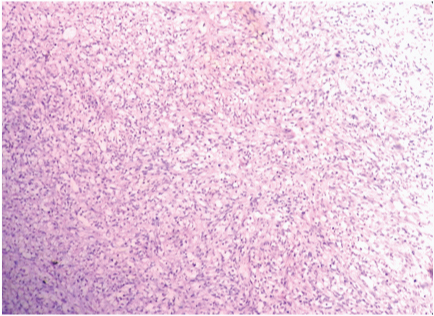
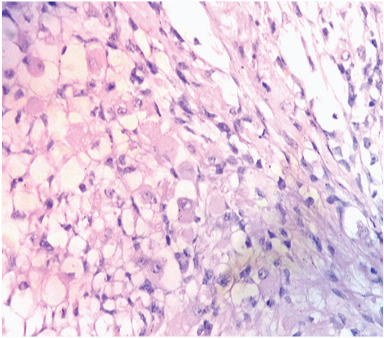
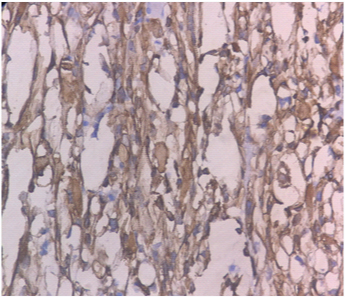
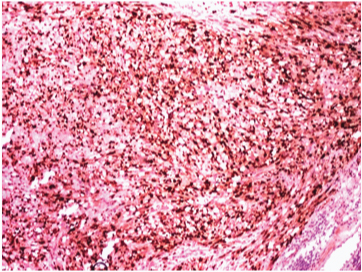
On gross examination, the specimen consisted of a grey-white soft tissue measuring 2.5x2x2cm and firm to touch. On microscopic examination, sections showed presence of a well circumscribed tumour consisting of highly cellular areas characterized by arrangement of neoplastic, moderately atypical spindle cells, arranged in intersecting fascicles [Table/Fig-1]. Numerous atypical mitoses were seen (12/10 HPF). This led to a suspicion of leiomyosarcoma or fibrosarcoma. However, on careful inspection, it was observed, that admixed with the spindle cells, were few polygonal cells having eccentric nuclei, prominent nucleoli and eosinophilic cytoplasm–rhabdomyoblasts [Table/Fig-2]. These cells were the most important clue to the diagnosis on light microscopy. Immunohistochemical staining of Desmin showed diffuse cytoplasmic positivity in the spindle cells as well as the polygonal cells, along with presence of cross striations in many neoplastic cells [Table/Fig-3]. Immunohistochemistry for Myogenin showed 4+ positivity in the tumour cell nuclei ([Table/Fig-4], sourced from Lal pathlabs). Because spindle cells constituted more than 50 % of the neoplastic population, a diagnosis of spindle cell variant of rhabdomyosarcoma was made. The patient was referred to a higher center for treatment, but chose not to seek medical help. Eventually, the patient came back to the outpatient department after two and a half months due to recurrence of the swelling. She was referred again, and was lost to follow-up.
Low power view showing fascicular arrangement of tumour cells. Few atypical mitoses are seen. (H&E x 100).
High power view shows presence of rhabdomyoblasts. (H&E x 400).
Immunohistochemical stain for Desmin showing positivity in the spindle cells, and cross striations. (DAB x 400).
Immunohistochemical stain for Myogenin showing intense positivity in the nuclei. (DAB x 400).
Discussion
Rhabdomyosarcomas are the most common soft-tissue malignancy in children and adolescents, comprising 51% of sarcomas reported every year according to data from the Surveillance, Epidemiology, and End Results (SEER) Program [1].
Rhabdomyosarcoma has three histologic subtypes; embryonal, alveolar and pleomorphic. The embryonal subtype, which is the most common, has three variants, Spindle cell, Botyroid and Anaplastic [2].
The spindle cell variant of embryonal rhabdomyosarcoma was first recognized as a rare entity having a male predilection, propensity for occurrence in the paratesticular, and head and neck regions, and a low malignant potential, in 1992 by German-Italian Cooperative Soft Tissue Sarcoma Study [3]. It accounts for 3-4.4 % of all subtypes of rhabdomyosarcoma [4], and has a better prognosis in children [5].
The tumours usually present as a gradually increasing painless, firm swelling [6]. The size of these lesions can vary considerably, with a range of 1.5-35 cm [7].
Histologically, the tumour is composed mainly of elongated spindle cells arranged in a fasciculated or storiform pattern, mimicking smooth muscle fibers [3]. Mitotic figures are easily appreciated, including atypical forms. Admixed with the spindle cells, is a second population of immature rhabdomyoblasts, usually comprising a small population of the tumour [8].
Spindle cell rhabdomyosarcoma (SC-RMS) has been classified into collagen rich and collagen poor forms. In the collagen book form, the tumour cells are separated by abundant collagen fibres. The collagen rich form on the other hand, is very cellular, comprising of spindle shaped cells in fascicles, without much intervening collagen. This form is more likely to be misdiagnosed as leiomyosarcoma on morphology [9]. The tumour in the present case is a collagen poor spindle cell variant of rhabdomyosarcoma.
The histopathological differentials are spindle cell neoplasms like leiomyosarcoma [5,9], fibrosarcoma, or a malignant peripheral nerve sheath tumour with heterologous differentiation (malignant triton tumour) [10].
The spindled tumour cells, the whorled arrangement, and positivity for certain markers like desmin, seen in both leiomyosarcomas and Spindle cell Rhabdomyosarcoma, may lead to a mistaken diagnosis of a smooth muscle tumour, in SC-RMS. A herringbone pattern may also be seen in an SC-RMS, causing a problem in differentiation from a fibrosarcoma. In both these situations, immunoreactivity for muscle specific markers like myogenin will help solve the problem. A desmoplastic melanoma with spindled cells may be differentiated from a SC-RMS, by demonstration of S-100 positivity. It may not always be possible to distinguish between SC-RMS and a malignant triton tumour [11] on the basis of morphology and Immunohistochemistry, because the latter may sometimes be negative for S-100. A diagnosis of neurofibromatosis may be helpful in this case [6].
In the present case, a diagnosis of leiomyosarcoma was first made because of the tumour cell morphology and fascicular arrangement. But the age of the patient, and the anatomic location of the neoplasm prompted us to look further. The recognition of rhabdomyoblasts on an H and E stained section, identification of cross striations on Desmin stain and finally, intense nuclear positivity for myogenin helped to confirm the diagnosis of an Embryonal Rhabhdomyosarcoma.
Pediatric spindle cell rhabdomyosarcomas have been shown to have a better prognosis compared with other rhabdomyosarcomas [9]. These tumours are locally aggressive, and this makes them difficult to excise completely in the head and neck region. In the paratesticular location however, these tumours are diagnosed earlier and are more amenable to removal. So, the prognosis is better in paratesticular tumours [5,9].
SC-RMS is currently being treated with protocols similar to other rhabdomyosarcomas. This is because they are rare, and not enough research has been done on an alternate therapeutic approach. The standard protocols include surgery, chemotherapy and radiation. In children, SC-RMS has a good prognosis, with a favorable response in more than 90% [3,5]. However, in adults, 5-year survival is reported to be about 53% by Stock et al., [12].
Conclusion
The spindle cell variant of embryonal rhabdomyosarcomas is a rare variant of embryonal subtype of Rhabdomyosarcoma, occurring more commonly in children and adolescents. Its low malignant potential and better prognosis in children makes the correct diagnosis crucial using combination of clinical features, histopathology and Immunohistochemistry.
[1]. Miller RW, Young JL, Novakovic B, Childhood cancerCancer 1995 75:395-405. [Google Scholar]
[2]. Parham DM, Barr FG, Embryonal rhabdomyosarcoma. In: Fletcher CDM, Unni KK, Mertens F, edsWHO Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone 2002 LyonIARC Press:146-49. [Google Scholar]
[3]. Cavazzana AO, Schmidt D, Ninfo V, Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcomaAm J Surg Pathol 1992 16(3):229-35. [Google Scholar]
[4]. Weiss SS, Goldblum JR, Rhabdomyosarcoma. In: Weiss SS, Goldblum JR edsEnzinger and Weiss’s Soft Tissue Tumours 2008 5th edPhiladelphiaElsevier:595-632. [Google Scholar]
[5]. Leuschner I, Newton WA, Schmidt D, Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region: a report of the Intergroup rhabdomyosarcoma studyAm Surg Pathol 1993 17(3):221-30. [Google Scholar]
[6]. Carroll SJ, Nodit L, Spindle Cell rhabdomyosarcoma. A brief diagnostic review and differential diagnosisArch Pathol Lab Med 2013 137:1155-58. [Google Scholar]
[7]. Nascimento AF, Fletcher CD, Spindle cell rhabdomyosarcoma in adultsAm J Surg Pathol 2005 29(8):1106-13. [Google Scholar]
[8]. Dehner LP, Skeletal system. In: Stocker JT, Dehner LP, Husain AN, edsPediatric Pathology 2011 3rd edPhiladelphiaLippincott Williams & Wilkins:1190-1270. [Google Scholar]
[9]. Newton WA, Gehan EA, Webber BL, Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification – an Intergroup rhabdomyosarcoma studyCancer 1995 76(6):1073-85. [Google Scholar]
[10]. Rubin BP, Hasserjian RP, Singer S, Spindle cell rhabdomyosarcomas (so- called) in adults: report of two cases with emphasis of the differential diagnosisAm J Surg Pathol 1998 22(4):459-64. [Google Scholar]
[11]. Stasik CJ, Tawfik O, Malignant peripheral nerve sheath tumour with Rhabdomyosarcomatous differentiationArch Pathol Lab Med 2006 130:1878-81. [Google Scholar]
[12]. Stock N, Chibon F, Binh MB, Adult type Rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosisAm J Surg Pathol 2009 33(12):1850-59. [Google Scholar]