Hepatosplenic T Cell Lymphoma in an Immunocompetent Female Diagnosed using Flow Cytometry: A Rare Clinical Entity
Pranav Dorwal1, Ritesh Sachdev2, Amit Pande3, Dharmendra Jain4, Bhawna Jha5, Vimarsh Raina6
1 Associate Consultant, Department of Pathology and Lab Medicine, Medanta The Medicity, Gurgaon, Haryana, India.
2 Senior Consultant, Department of Pathology and Lab Medicine, Medanta The Medicity, Gurgaon, Haryana, India.
3 Junior Scientist, Department of Pathology and Lab Medicine, Medanta The Medicity, Gurgaon, Haryana, India.
4 Scientist, Department of Pathology and Lab Medicine, Medanta The Medicity, Gurgaon, Haryana, India.
5 Associate Consultant, Department of Pathology and Lab Medicine, Medanta The Medicity, Gurgaon, Haryana, India.
6 Director, Department of Pathology and Lab Medicine, Medanta The Medicity, Gurgaon, Haryana, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Pranav Dorwal, Department of Pathology & Laboratory Medicine, UG Floor, Clinical Labs Medanta The Medicity, Sector 38, Gurgaon-122001, Haryana, India.
E-mail: dorwal@rediffmail.com
Hepatosplenic T-cell lymphoma is a rare haematopoietic malignancy that comprises less than 1% of Non-Hodgkin lymphomas. We are reporting a case of a 26-year-old female, who presented with pallor, weight loss, jaundice, pancytopenia and hepatosplenomegaly. The bone marrow examination showed infiltration by lymphoid cells. These cells on flow cytometric evaluation showed the phenotype of hepatosplenic T cell lymphoma. The cells were positive for CD3, CD8, CD56 and TCR γδ and negative for CD5, CD4, CD8, CD16, CD57, TCRαβ along with B cell markers. This case is reported for being a rare clinical entity and its presence in an immunocompetent female making it rarer.
Immunophenotyping, Cluster of differentiation, Hepatosplenomegaly, Pancytopenia
Case Report
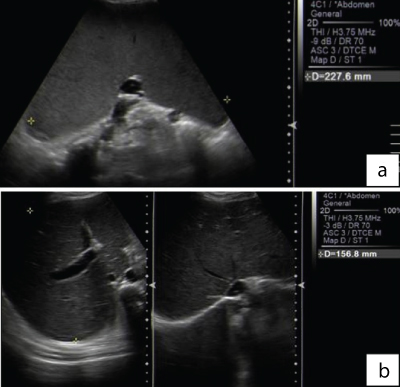
A 26-year-old female patient presented with a history of pallor, weight loss, jaundice and pancytopenia for which she was evaluated by a general practitioner and found to have hepatosplenomegaly. The patient was referred to our hospital and on initial evaluation was found to have deranged liver functions. The various laboratory parameters of the patient were: Haemoglobin: 9gm/dl, WBC count: 1600/μl, platelets: 80000/μl total serum bilirubin: 15.4 mg/dl, conjugated bilirubin: 10.25 mg/dl, SGOT: 750 U/L, SGPT: 144 U/L, GGT: 214 U/L, ALP: 975 U/L and LDH: 4659 U/L. Renal function tests were normal. The viral markers (for hepatitis B, hepatitis C and HIV) and autoimmune profile (ANA and ANCA) was unremarkable. The ultrasonographic examination of the abdomen revealed mild hepatomegaly (15.6 cm; midclavicular line, craniocaudal length) and massive splenomegaly (22.7 cm; superior to inferior axis) [Table/Fig-1]. The PET scan revealed splenomegaly with mildly increased FDG uptake.
(a) Spleen is markedly enlarged in size (22.7cm) and shows normal echotexture. No focal lesion is seen. B: Liver is mildly enlarged in size (15.6cm) and shows normal echotexture. No obvious focal lesion is seen. No intra hepatic biliary duct dilatation. CBD is normal.
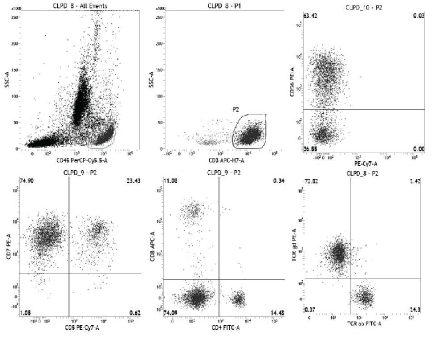
In view of the pancytopenia, bone marrow aspiration was performed. It showed presence of atypical lymphoid cell infiltration. The flow cytometry was performed in order to evaluate the immunopehnotype of the atypical lymphoid cells. A comprehensive panel comprising of T, B and NK cell markers was put up. The lymphoid cells were predominantly T cells (CD3 +). They were gated and further evaluated, and were found to show abnormal T cell expression. This included lack of both CD4 and CD8 along with lack of CD5 while CD7 positivity was present [Table/Fig-2]. They were also positive for CD56, and negative for CD16 and CD57 (All antibodies from BD Biosciences, San Jose, CA, USA). The T cell receptor analysis revealed that the abnormal cells were positive for TCR gamma-delta, while scanty of cells showed TCR alpha-beta expression (normal residual T cells). All the B cell markers were negative.
The flow cytometry performed showed predominance of lymphoid cells. The entire population of lymphoid cells were gated (P1, upper-left). These gated population composed nearly entirely of CD3+ cells, which were gated (P2, upper-middle). The gated CD3+ cells (P2) were CD56 positive (upper-right), CD7 positive (lower-left), CD5 negative (lower-left), CD4 negative (lower-middle), CD8 negative (lower-middle), and expressed TCR γδ (lower-right).
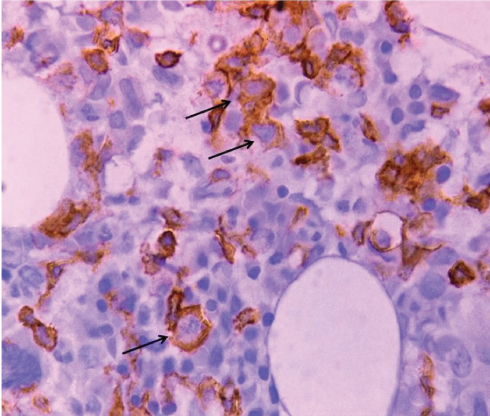
The immunophenotype in light of the clinical and radiological findings was compatible with Hepatosplenic T cell Lymphoma. The bone marrow biopsy also showed infiltration by T cells as identified on immunohistochemistry for CD3 [Table/Fig-3]. Liver biopsy was not performed, as the haematologist decided against it considering the morbidity associated with it. He had sufficient evidence in form of bone marrow aspiration, biopsy, flow cytometry and radiological findings to make a definitive diagnosis and start the treatment. The patient was started on modified CHOP, and this resulted in fall in serum bilirubin, normalization of liver enzymes and reduction in spleen size by 60%. There was also considerable improvement in hemoglobin which reached 12.5gm/dl, along with marked improvement in the B symptoms. There was persistent disease on bone marrow examination and the patient was offered autologous stem cell transplant. However, the patient and her family refused the same, citing financial reasons. They were apprised of the poor prognosis with the persistent disease. The patient has since been lost to follow-up.
Immunohistochemical staining of the bone marrow biopsy for CD3 (IHC, 400x). arrows signify positivity for CD3.
Discussion
Hepatosplenic T-cell Lymphoma (HSTL) is a rare T cell neoplasm that is known to occur mainly in the young males [1]. This entity was first described by Farcet, Gaulhard et al., in 1990 and since then only a limited number of cases of this entity have been reported [2]. As the population of cells in the bone marrow can be very low, it is important to identify the abnormal expression of T cell markers in this disease entity. The hepatosplenomegaly is seen due to the sinusoidal involvement by the neoplastic T cells. The disease has an aggressive clinical course with extremely poor prognosis. Initially it was named as Hepatosplenic gamma-delta T cell Lymphoma, but was later changed to Hepatosplenic T cell Lymphoma in the WHO classification of 2008 [3]. It is considered that HSTL cells originate from peripheral γδ cytotoxic memory T-cells of the innate immune system, in the spleen, as the normal γδ T-lymphocytes have a special homing tendency for spleen.
In one of the reviews, it was found to be five times more common in males as compared to females [4]. A quarter of the patients diagnosed with HSTL have a history of immune suppression, including those in post-transplant setting, or those treated with azathioprine and infliximab for Crohn’s disease [5]. The thorough review conducted by Kotlar DS et al., stated that HSTL was seen mostly in inflammatory bowel disease patients who had received thiopurines for 2 years or more [6]. The common presentation is that of thrombocytopenia, along with leucopenia and anemia. HSTL usually show mild elevation of liver enzymes which is due to the liver cell damage and cases with even fulminant hepatic failure have been reported [7]. The elevated bilirubin levels are considered to be due to periportal adenopathy, which could be the cause in the current case too [8]. The most common sites of involvement include spleen, liver and bone marrow. This was the classical presentation in our case too. Peripheral blood involvement is rare and usually late in the course of the disease. The typical immunophenotype of this entity is CD2+, CD3+, CD4-, CD5-, CD7+, CD8-, with expression of TCR γδ; however few cases may show expression of TCRαβ [9]. Our case showed this classical immunophenotype. Genetic profiling of this lymphoma usually shows rearrangement for TRG genes. Isochromosome 7q and trisomy 8 have been known to be present in most of the cases. This was not performed in our case as the patient was poor and it was not feasible for her to undertake further investigations. The 2005 Society for Hematopathology and European Association of Hematopathology Workshop said that the tumour cells tend to infiltrate the sinusoids of the splenic red pulp, liver and bone marrow [10]. It also said that the portal and periportal infiltration is unusual in liver. There is no specifically designated therapy for HSTL cases and a variety of therapies have been used including steroids, alkylating agents, purine analogs, splenectomy and high dose chemotherapy with allogenic stem cell transplant [4]. The prognosis of HSTL is extremely poor with the median survival being less than 2 years [3].
Conclusion
This case is being reported for being a rare clinical entity and its presence in an immunocompetent female making it rarer.
[1]. Balhadj K, Reyes F, Farct JP, Tilly H, Bastard C, Angonin R, Hepatosplenic γδ T-cell Lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patientsBlood 2003 102:4261-19. [Google Scholar]
[2]. Farcet JP, Gaulard P, Marolleau JP, Le Couedic JP, Henni T, Gourdin MF, Hepatosplenic T-cell Lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma deltaBlood 1990 75(11):2213-19. [Google Scholar]
[3]. Gaulard P, Jaffe ES, Krenacs L, Macon WR, Hepatosplenic T-cell lymphomaWHO classification of tumours of haematopoietic and lymphoid tissues 2008 LyonIARC:292-293. [Google Scholar]
[4]. Weidmann E, Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990Leukmia 2000 14(6):991-97. [Google Scholar]
[5]. Mackey AC, Gren L, Liang LC, Dinndorf P, Avigan M, Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel diseaseJ Pediatr Gastroenterol Nutr 2007 44:265-67. [Google Scholar]
[6]. Kotlyar DS, Osterman MT, Diamond RH, Porter D, Blonski WC, Wasik M, A systematic review of factors that contribute to Hepatosplenic T-cell lymphoma in patients with inflammatory bowel diseaseClin Gastroenterol & Hepatol 2011 9(1):36-41. [Google Scholar]
[7]. Lettieri CJ, Berg BW, Clinical features of non-Hodgkin’s lymphoma presenting with acute liver failure: a report of five cases and review of published experienceAm J Gastroenterol 2003 98:1641-46. [Google Scholar]
[8]. Lu CL, Tang Y, Yang QP, Wang M, Zaho S, Bi CF, Hepatosplenic T-cell lymphoma: clinicopathologic, immunophenotypic and molecular characterization of 17 Chinese casesHuman Pathol 2011 42:1965-78. [Google Scholar]
[9]. Alsohaibani FI, Abdulla MA, Fagih MM, Hepatosplenic T cell lymphomaIndian J Haematol Blood Transfus 2011 27(1):39-42. [Google Scholar]
[10]. Vega F, Medeiros J, Gaulard P, Hepatosplenic and other γδ T-cell lymphomasAm J Clin Pathol 2007 127:869-80. [Google Scholar]