Endobronchial Inflammatory Myofibroblastic Tumour-A Case Report
Vishnukanth Govindaraj1, Debasis Gochhait2, Balla Nagamalli Kumar3, Suryakala Narayanasami4
1 Assistant Professor, Department of Pulmonary Medicine, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), D Nagar, Puducherry, India.
2 Assistant Professor, Department of Pathology, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), D nagar, Puducherry, India.
3 Junior Resident, Department of Pulmonary Medicine, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), D Nagar, Puducherry, India.
4 Senior Resident, Department of Pulmonary Medicine, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), D Nagar, Puducherry, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Vishnukanth Govindaraj, No:3, Rajaya Street, Radhakrishnan Nagar, Ariyankuppam, Puducherry-605007, India.
E-mail: vishnu1429@yahoo.com
Lung malignancies are on the rise and sadly present at an advanced stage. Fiberoptic bronchoscopy is used for staging as well as in diagnosis of lung malignancies. However, not all endobronchial growth are malignant. Inflammatory Myofibroblastic Tumour (IMT) is one of the rare tumours of the lung. A controversy regarding the benign versus malignant nature of the tumour is still ongoing. The management of these tumours can be challenging because there are no established treatment protocols. Although IMT most commonly arises from lung, endobronchial presentation is very rare. We report a case of endobronchial presentation of IMT and discuss about its aetiology and treatment options.
Fiberoptic bronchoscopy, Inflammatory pseudotumour, Lung malignancies, Smooth muscle antigen
Case Report
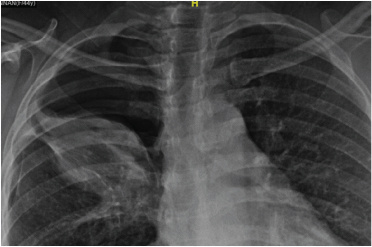
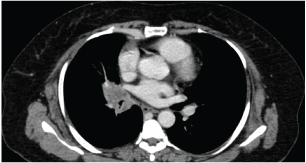
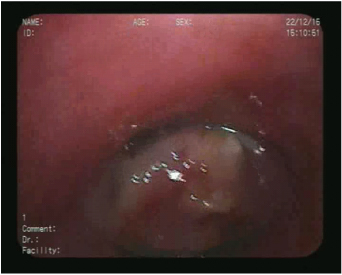
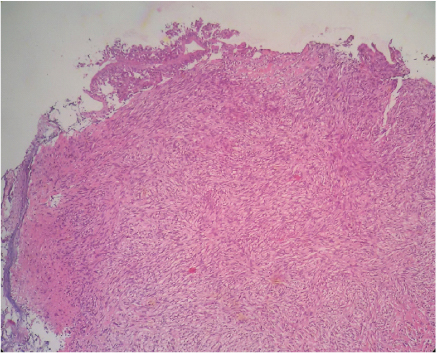
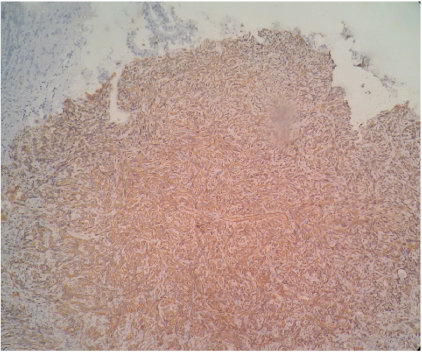
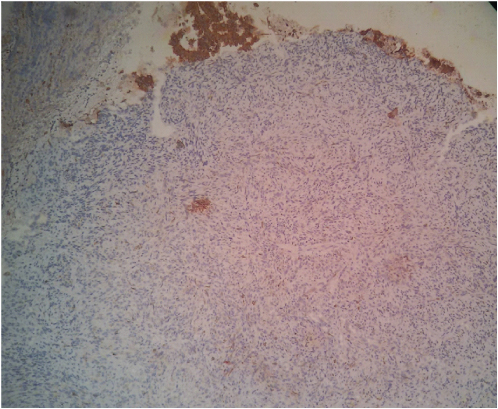
A 55-year-old postmenopausal housewife presented with three months history of cough with expectoration. She also had occasional right sided chest pain and scanty haemoptysis on a couple of occasion. She was already evaluated elsewhere in detail for tuberculosis. She had no co-morbid illness. Upon presentation to our institute, she was stable and maintained saturation at room air. Her respiratory system examination revealed inspiratory crackles in the right mammary region. Her blood counts, renal function tests, liver function tests were normal. Chest X-ray showed a non-homogenous opacity in the right upper zone [Table/Fig-1]. Computed Tomography (CT) scan of the chest was performed which showed a right hilar mass with narrowing of bronchus intermedius [Table/Fig-2] possibility of right bronchogenic carcinoma with post obstructive pneumonitis was considered. CT guided biopsy was obtained from the right hilar mass which was inconclusive. Patient developed minimal pneumothorax post biopsy which was managed conservatively. Fiber optic bronchoscopy was done and it showed a narrowed bronchus intermedius with an endobronchial polypoid lesion of 5-7mm [Table/Fig-3]. A biopsy of the lesion was done and sent for histopathological examination. The biopsy showed focal intact lining epithelium with a spindle cell lesion in the sub epithelial area. The spindle cells were arranged in a storiform pattern and were intermixed with a mixed inflammatory infiltrate comprising of lymphocytes, many eosinophils and few histiocytes [Table/Fig-4,5]. There was no significant atypia or mitosis in the spindle cell proliferation. On Immunohistochemistry (IHC) these spindle cells were positive only for Smooth Muscle Actin (SMA) and negative for Pan Cytokeratin (Pan Ck), S-100, Desmin, Anaplastic lymphoma kinase (ALK) and CD-30 [Table/Fig-6,7]. In view of the diffuse positivity for SMA in the spindle cells and mixed inflammatory infiltrate, the lesion was diagnosed as Inflammatory myofibroblastic tumour. Metastatic workup was negative. The patient was explained about the need for surgical resection of the mass but she declined due to personal reasons. Currently the patient is on regular follow-up and her condition has not worsened till last contact.
Chest x ray showing a non-homogenous opacity in the right upper zone. A thin rim of pneumothorax is also seen.
CT thorax showing an endobronchial growth in the right bronchus intermedius.
Fiberoptic bronchoscopy showing an endobronchial polypoidal growth in the right bronchus intermedius.
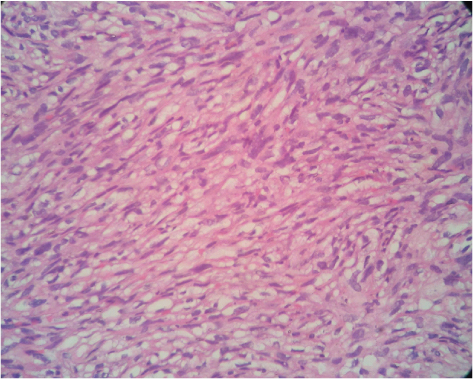
Haematoxylin and eosin stain section (200X magnification) shows spindle cell proliferation in the sub epithelial portion. The spindle cells are intermixed with scattered lymphocytes and eosinophils.
Haematoxylin and eosin stain section (400 X magnifications) shows spindle cell proliferation in a repetitive storiform pattern. The spindle cells are intermixed with lymphocytes and eosinophils.
Immunohistochemistry stain for Smooth muscle actin (SMA - 200 X magnifications) shows diffuse positivity in the cytoplasm of the spindle cells.
Immunohistochemistry stain for Pan Cytokeratin (Pan CK - 200 X magnifications) shows diffuse positivity in the cytoplasm of the surface epithelial cells but negative in the underlying spindle cells.
Discussion
Inflammatory Myofibroblastic Tumour (IMT) also known as plasma cell granuloma or inflammatory pseudotumour is one of the rare tumours of the lung [1]. IMTs arise from various anatomic sites, predominantly from lung followed by orbit, retroperitoneum and abdominopelvic organs [2]. IMT was first observed and described by Brunn in 1939 [3]. Due to the presence of variable cellular components, these tumours were also referred as plasma cell granuloma, inflammatory myofibroblastic tumour, histiocytoma, fibroxanthoma, xanthogranuloma and plasma cell histiocytoma [4]. Umiker and Iverson coined the term inflammatory myofibroblastic tumour as these lesions mimic malignant lesions both clinically and histopathologically [5]. IMT accounts for 0.04%-1% of all lung tumours [6]. The exact aetiology of IMT is unknown. However infections with mycobacteria, Epstein barr virus, actinomyces, nocardia, mycoplasma, herpes simplex virus have been implicated in the causative process. A possibility of autoimmune process as a possible aetiology is also proposed [3,7]. Role of autoimmune process in the pathogenesis of IMT is supported by the fact that Immunoglobulin G4 – related sclerosing disease resembles IMT [8].
IMT tends to occur in persons under 40 years of age with both sexes affected equally [1]. Our patient was a 55-year-old house wife. Majority of the patients are asymptomatic. Patients with endobronchial lesions have symptoms of cough, haemoptysis, dyspnoea and chest pain. There is no specific radiological sign of IMT. Agrons et al., analysed radiological findings of pulmonary IMT. Solitary nodule was the common finding in about 90% of the CT scans. Multiple nodules were seen in about 5% and infiltration of the hilum, mediastinal and airways were seen in 16% of the patients. Endobronchial presentation is rare and accounts for less than 12% of the reported cases [9]. Cases of local invasion and metastatic spread of IMT have also been reported particularly in extra pulmonary lesions [10].
The diagnosis of IMT is incidental. As there are no specific clinical or radiological sign for IMT, diagnosis is usually not suspected until a histopathological testing is done. Tissues obtained via computed tomography–guided fine-needle aspiration or trucut biopsy may not reveal the exact diagnosis. Most of the time, the whole resected specimen may have to be examined to confirm the diagnosis. Endobronchial lesions can be biopsied under bronchoscopy guidance. In our case, the initial percutaneous biopsy was inconclusive. On histopathological examination IMT is composed of mesenchymal spindle cells and variable inflammatory including plasma cells, histiocytes, lymphocytes. Depending on the predominant cell type, various subclassification of IMT has been proposed.
Matsubara et al., classified IMT into three groups based on predominant cell type and histologic properties: 1) Organized pneumonia formed by gradual healing of the intra alveolar exudation; 2) Fibrous histiocytoma formed by storiform proliferation of plasma cell and lymphocyte aggregates; 3) lymphoplasmocytic type formed by the aggregation of both plasma cell and lymphocytes [6]. Our patient had a storiform arrangement of spindle cells. Due to its multicellular mixture, IMT has to be carefully differentiated from a number of conditions. The differential diagnosis includes malignant lymphoma, lymphoid hyperplasia, pseudolymphoma, plasmacytoma, malignant fibrous histiocytoma, sarcomatoid carcinoma of the lung, sclerosing haemangioma, sarcoma, and/or nodular chronic pneumonitis [11]. Translocations involving anaplastic lymphoma kinase (ALK) gene or ALK protein expression in IMT have been reported, especially in younger patients. The detection of this gene rearrangement strengthens the diagnosis of IMT. However, its absence does not exclude the diagnosis [2]. Demonstration of ALK positivity is a relatively good prognostic variable.
The possibility of IMT being a low grade malignancy is supported by studies showing clonal chromosomal abnormalities, chromosomal rearrangements involving ALK receptor tyrosine kinase locus region and DNA aneuploidy. In 1994, WHO has classified IMT as tumours of intermediate malignant potential due to a tendency for local recurrence and its small risk for distant metastasis [12].
Treatment of IMT is challenging as there are no established treatment protocols. Surgical resection is the treatment of choice for IMT. In patients undergoing radical resection, recurrences are rare. Retroperitoneal and intraperitoneal IMTs tend to invade local structures making complete resection challenging. Completely resected lesions <3cm in diameter are associated with a five year survival of more than 90%.
Chemotherapy and radiotherapy have found be less effective in IMT. Corticosteroids have been found to be effective in IMT arising from orbit. Studies using anti-tumour necrosis factor-α binding antibodies are promising [13]. Incidences of successful treatment of retroperitoneal IMT with a combination of non-steroidal anti-inflammatory drugs and platinum based chemotherapy have been reported [14]. Crizotinib has found to be useful in IMT exhibiting ALK rearrangement [10]. Few cases of spontaneous remission of IMT have also been reported [15].
Conclusion
In conclusion, IMT are tumours with a possible malignant potential and excellent prognosis if detected and resected early. Though rare, IMT should be considered in the differential diagnosis of a lung tumour particularly if slow growing.
[1]. Demirhan O, Ozkara S, Yaman M, Kaynak K, A rare benign tumour of the lung: Inflammatory myofibroblastic tumour-case reportRespiratory Medicine Case Reports 2013 8:32-35. [Google Scholar]
[2]. Jacob SV, Reith JD, Kojima AY, Williams WD, Liu C, Duckworth LV, An unusual case of systemic inflammatory myofibroblastic tumour with successful treatment with ALK-InhibitorCase Reports in Pathology 2014 2014:470340 [Google Scholar]
[3]. Kaitoukov Y, Rakovich G, Trahan S, Grégoire J, Inflammatory pseudotumour of the lungCan Respir J 2011 18(6):315-17. [Google Scholar]
[4]. Dishop MK, Warner BW, Dehner LP, Successful treatment of inflammatory myofibroblastic tumour with malignant transformation by surgical resection and ChemotherapyJournal of Pediatric Haematology/Oncology 2003 25(2):153-58. [Google Scholar]
[5]. Patel SB, Shah DM, Goswami KG, Shah SR, Shah PM, Inflammatory pseudotumour of lung, a case report and reviewInd J Radiol Imag 2006 16(1):117-18. [Google Scholar]
[6]. Matsubara O, Tan-Liu NS, Kenney RM, Mark EJ, Inflammatory pseudotumours of the lung: progression from organizing pneumonia to fibrous histiocytoma or to a plasma cell granuloma in 32 casesHum Pathol 1988 19:807-14. [Google Scholar]
[7]. Palaskar S, Koshti S, Maralingannavar M, Bartake A, Inflammatory myofibroblastic tumour of the jaw- a case reportContemp Clin Dent 2011 2(4):274-77. [Google Scholar]
[8]. Zen Y, Kitagawa S, Minato H, IgG4-positive plasma cells in inflammatory pseudotumour (plasma cell granuloma) of the lungHum Pathol 2005 36:710-17. [Google Scholar]
[9]. Agrons GA, Rosado-de Christenson ML, Kirejcyzk WM, Conran RM, Stocker JT, Pulmonary inflammatory pseudotumour radiologic featuresRadiology 1998 206:511-18. [Google Scholar]
[10]. Inadomi K, Kumagai H, Takayoshi K, Ariyama H, Kusaba H, Nishie A, Successful combination chemotherapy for metastatic inflammatory myofibroblastic tumour: A case reportOncology letters 2015 10:2981-85. [Google Scholar]
[11]. Sakuraia H, Hasegawab T, Watanabea S, Inflammatory myofibroblastic tumour of the lungEuropean Journal of Cardio-thoracic Surgery 2004 25:155-59. [Google Scholar]
[12]. Biselli R, Boldrini R, Ferlini C, Boglino C, Inserra A, Bosman C, Myofibroblastic tumours: neoplasias with divergent behaviour. Ultrastructural and flow cytometric analysisPathology Research and Practice 1999 195(9):619-32. [Google Scholar]
[13]. Germanidis G, Xanthakis I, Tsitouridis I, Regression of inflammatory myofibroblastic tumour of the gastrointestinal tract under infliximab treatmentDig Dis Sci 2005 50:262-65. [Google Scholar]
[14]. Tao Y-L, Wang Z-J, Han J-G, Wei P, Inflammatory myofibroblastic tumour successfully treated with Chemotherapy and nonsteroidals: A case reportWorld J Gastroenterol 2012 18(47):7100-03. [Google Scholar]
[15]. Mandelbaum I, Brashear RE, Hull MT, Surgical treatment and course of pulmonary pseudotumour (plasma cell granuloma)J Thorac Cardiovasc Surg 1981 82:77-82. [Google Scholar]