Mediastinal masses in children comprises of a heterogeneous group of tumours. In such cases, biopsy and histological analysis are mandatory for planning of treatment. We have reported an unusual aetiology for a mediastinal mass in a young boy presenting with features of Superior Vena Caval Obstruction (SVCO) who also had marked blood and marrow eosinophilia mimicking Chronic Eosinophilic Leukaemia (CEL). We have also discussed the differential diagnoses of mediastinal tumours with hyper-eosinophilia and possible therapeutic implications.
Chest mass, Histiocytosis, Lymphadenopathy, Mediastinal tumour, Superior Vena Caval Obstruction
Case Report
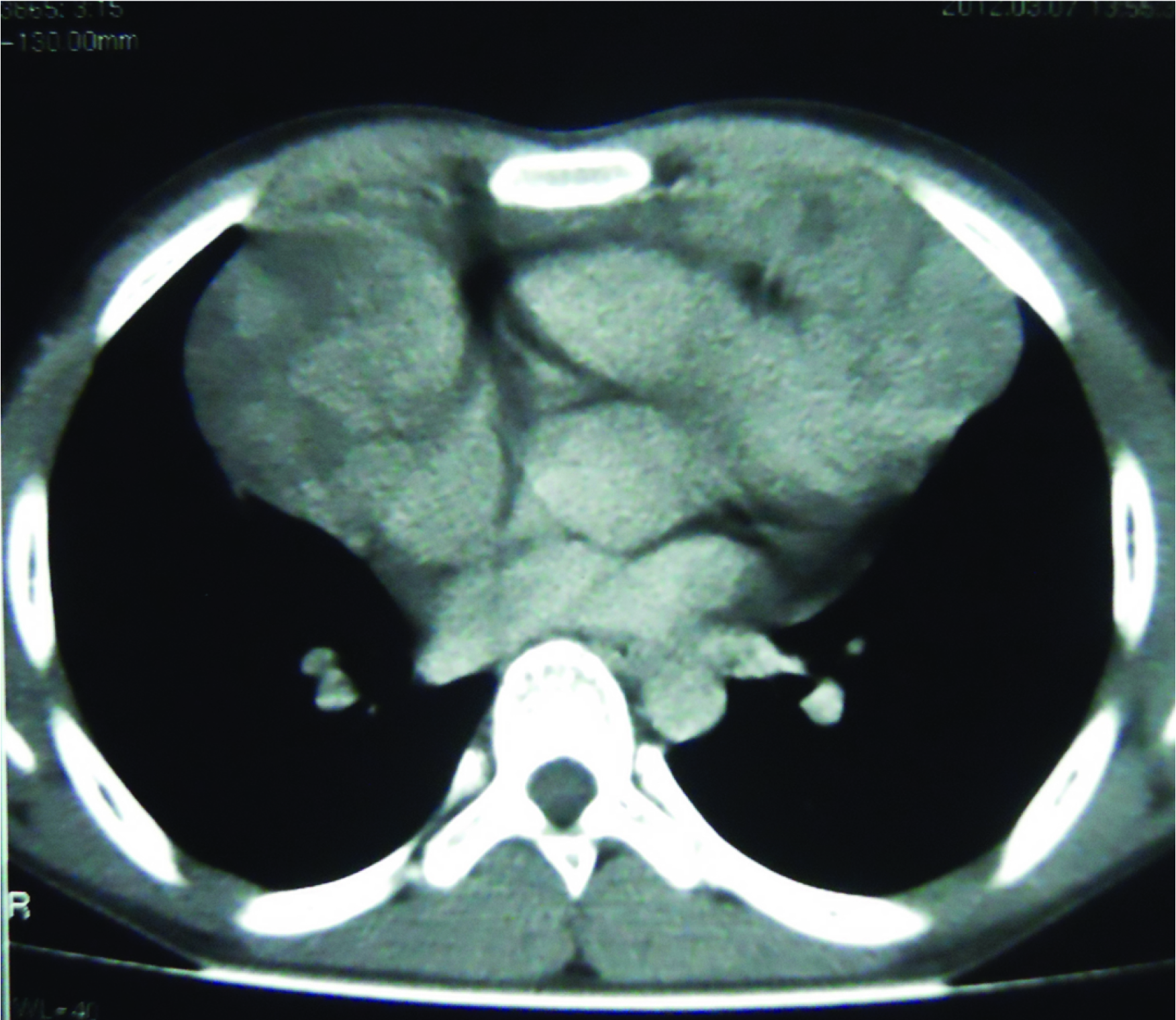
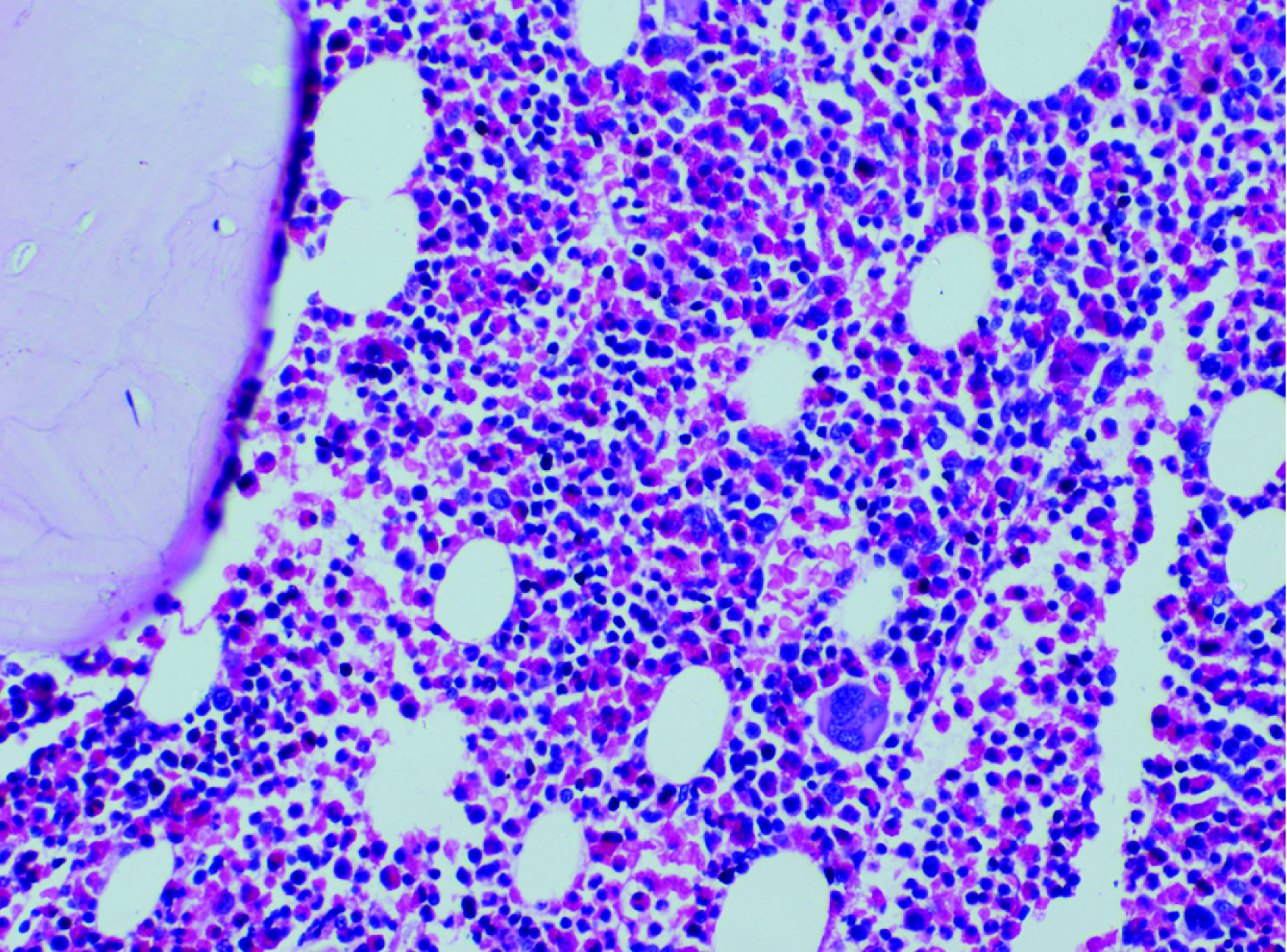
A 15-year-old boy presented with left tonsillar enlargement for 3 months associated with non productive cough and dyspnea of 15 days duration, aggravated in supine position. There was no history of fever or any significant loss of weight. On examination he had features of Superior Vena Caval Obstruction (SVCO) and right supraclavicular lymphadenopathy. Lymph nodes were firm and nontender. Laboratory parameters showed Hb of 13.6 g/dl, TC -31,600/mm3, DC-N11%, L10%, E-79% (AEC-24,964/mm3), platelet - 3.53 lac/mm3, peripheral smear showed 80% eosinophils. CT chest [Table/Fig-1] revealed extensive mediastinal lymphadenopathy, USG abdomen and skeletal survey were normal. The differential diagnosis considered were lymphoblastic/Hodgkin’s lymphoma or Chronic Eosinophilic Leukaemia (CEL) with extra-medullary deposits of eosinophils. Bone marrow aspiration/biopsy showed 59% eosinophils with dysplastic changes [Table/Fig-2] and karyotype was normal. In view of the eosinophilia, RT-PCR for FIP1L1-PDGFRα was done on the peripheral blood eosinophils which were negative. Causes of secondary eosinophilia were also excluded.
CT thorax showing a large well defined lobulated heterogenous enhancing mass lesion in anterior mediastinum.
H&Ex 400 Section from bone marrow showing sheets of eosinophils.
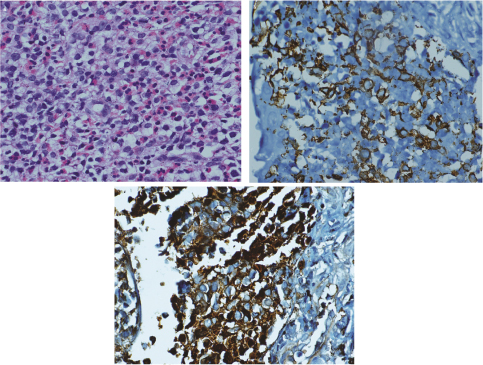
Biopsy of the mediastinal mass [Table/Fig-3a-c] showed infiltration by sheets of eosinophils admixed with linear cords of small cleaved cells which were LCA +ve, S100 +ve, CD1a +ve, CD25 +ve, CD30 -ve, CD3 -ve, and CD5 -ve. A final diagnosis of Langerhans cell histiocytosis (single system involvement -lymph node) with hyper-eosinophilia was made on the basis of immunohistochemistry on the biopsy sample of mediastinal mass and eosinophilia in blood smear.
(a) H&Ex400 Section from mediastinal mass showing sheets of elongated histiocytes admixed with eosinophils. (b) IHCx400 Histiocytes are CD1a positive. (c) IHCx400 histiocytes are S100 positive.
Our patient received local radiotherapy to mediastinum (24Gy in12 fractions). As systemic therapy he received steroids for four weeks and weekly vinblastine for six weeks followed by maintenance therapy with oral 6-Mercaptopurine for six months. CT chest after two months showed significant regression of the mediastinal mass and he also had resolution of the peripheral blood eosinophilia. He is in clinical, haematological and radiological remission for the last two years.
Discussion
Mediastinal masses in children are a very heterogeneous group of tumours- lymphoma, thymoma, germ cell tumours and bronchial or oesophageal cysts being the most common [1]. Langerhans Cell Histiocytosis (LCH) is a clonal, neoplastic proliferation of Langerhans cells, which are a normal constituent of the skin, lung, lymph node, and thymus. When LCH affects in organs such as skin, bone, lymph node and pituitary gland are easier to treat and is termed as low risk. LCH when affects Spleen, Liver and Bone marrow is difficult to treat and is termed as high risk. In most cases, LCH presents in older children and adults as unifocal disease that typically affects bone (skull, femur, pelvic bones) and less commonly lymph node, skin, or lung. However, it can also present as systemic disease with liver, spleen, lungs, bone marrow, endocrine, gastro-intestinal or central nervous system involvement [2]. In a series of 238 patients with LCH [3], 153 patients had unifocal disease, the most common site being a long or flat bone. Other common sites were lung, lymph nodes, skin and submandibular/parotid gland. Our patient presented with single system involvement of the mediastinal lymphnodes and staging work up did not reveal any other site of disease.
Fahrner et al., reported for the first time LCH originating from the mediastinum in an adult with a mediastinal tumour infiltrating the chest wall [1]. The patient received chemotherapy for LCH and at six months had significant regression of the chest wall mass. The authors concluded that in patients with mediastinal tumours, a histological diagnosis by excision biopsy is essential to plan adequate therapy.
Lee et al., reported a case of unifocal LCH of the thymus without clinical features of myasthenia gravis, presenting as an anterior mediastinal mass in a middle-aged woman with a prior history of resected leiomyosarcoma [4]. Thymic involvement by LCH occurs typically in children and as part of a systemic process. Gilcrease et al., reviewed seven cases of localized LCH in the thymus all of whom were infants or young children (4 patients) or young adults (3 patients) [5]. Interestingly, the young adults all presented with myasthenia gravis, and LCH was diagnosed at the time of thymectomy. Our patient also presented with an anterior mediastinal mass but the origin was from mediastinal lymph nodes.
LCH has been described co-existing with a number of malignancies, including non-Hodgkin and Hodgkin lymphoma [6] and adenocarcinoma of the lung and breast [7]. It is uncertain if this represents a genuine biological relationship or is coincidental. Egeler et al., reviewed 91 cases of LCH associated with malignant neoplasms in literature [7]. Most of these (67%) were associated with lymphoma or leukaemia; 12% were associated with solid tumours other than lung carcinomas, including 2 sarcomas, both arising from bone. Interestingly, the LCH frequently preceded the malignant neoplasm (in 78%). The authors postulated that these tumours could have arisen secondary to the radiation therapy used to treat LCH. In our patient also, close follow-up is warranted to detect the emergence of any second malignancy.
LCH with hyper-eosinophilia
Myeloid and lymphoid neoplasms with PDGFR and FGFR1 abnormalities can be associated with eosinophilia [8], which is clinically relevant as these neoplasms respond well to imatinib. Therefore neoplasms accompanied by persistent eosinophilia should be evaluated for PDGFR rearrangements. Other specific karyotypic abnormalities reported in eosinophilia associated with haematologic malignancies includes (5;14) (q31;q32) in ALL, 16q22 abnormalities in M4Eo subtype of AML and t(8;13)(pll;qll) translocation in patients with a rare syndrome of T-lymphoblastic lymphoma, eosinophilia, and myeloid hyperplasia [9].
The differential diagnosis of a mediastinal mass with hyper-eosinophilia which we considered in our patient was lymphoma/acute leukaemia or CEL with extra-medullary deposits of eosinophils in lymph nodes. RTPCR for FIP1L1-PDGFRα was negative and cytogenetics was normal, and other causes of secondary eosinophilia were excluded, hence the final diagnosis was LCH with hyper-eosinophilia. Since the eosinophilia in our patient resolved with treatment of LCH and since many of the agents used in our patient are effective in the treatment of CEL, we cannot be certain if the eosinophilia in our patient was secondary to LCH or of different aetiology.
Chen et al., in their study on patients with LCH reported that in approximately one quarter of the cases there were clonal rearrangements of the T-cell receptor gamma gene, immunoglobulin heavy chain and kappa light chain genes [10]. It may be postulated that a clonal T-cell population may be responsible for secreting cytokines like IL-3 and 5 which contribute to increased eosinophil proliferation in the marrow has been described in patients with the lymphocytic variant of Hypereosinophilic syndrome/CEL.
There are rare reports in literature of LCH presenting with hyper-eosinophilia. Ohnishi et al., reported for the first time a patient of CEL with FIP1L1–PDGFRα fusion that evolved post chemotherapy from LCH with reactive eosinophilia [11]. The patient was a young male initially diagnosed with multi-focal LCH of the bone and eosinophilia. Despite several courses of multi-drug chemotherapy for LCH, dysplastic eosinophils appeared in peripheral blood and marrow one year later. RT-PCR on bone marrow revealed the FIP1L1–PDGFRα fusion gene, which is frequently found in CEL. FIP1L1–PDGFRα was also analysed in the bone biopsy taken prior to chemotherapy and was negative suggesting a clonal evolution of eosinophils in this patient. Administration of imatinib in this patient caused transient remission of eosinophilia, but eosinophilia recurred 2 months later. Although the mechanisms by which FIP1L1–PDGFRα is generated are largely unknown, chemotherapy for the treatment of LCH might have caused FIP1L1–PDGFRα mutation in eosinophilic precursor cells in this patient. Therefore it would be prudent to monitor our patient for development of CEL in the future.
Our patient was a case of single system involvement (Lymphnodes) making it a minor category but as he had features of SVCO which is an emergency so early resolution of the mass was warranted hence he was treated with radiotherapy and chemotherapy with vinblastine and steroids [12]. He remained in remission even after 2 years of completed treatment.
Conclusion
Langerhans cell histiocytosis (LCH) should be considered in the differential diagnosis of a mediastinal mass in children. Rarely LCH can be associated with hyper-eosinophilia mimicking CEL but its prognostic and therapeutic implications are unknown. Such patients should be monitored for clonal evolution to CEL in the future.
[1]. Fahrner R, Hoksch B, Gugger M, Schmid AR, Langerhans cell histiocytosis as differential diagnosis of a mediastinal tumorEuropean Journal of Cardio-thoracic Surgery 2008 33:515-16. [Google Scholar]
[2]. Allen CE, Ladisch S, McClain KL, How i treat langerhans cell histiocytosisBlood 2015 126(1):26-35. [Google Scholar]
[3]. Lieberman PH, Jones CR, Steinman RM, Langerhans cell (eosinophilic) granulomatosis: A clinicopathologic study encompassing 50 yearsAm J Surg Pathol 1996 20:519-52. [Google Scholar]
[4]. Lee BH, George S, Kutok JL, Langerhans cell histiocytosis involving the thymusArch Pathol Lab Med 2003 127:e294-97. [Google Scholar]
[5]. Gilcrease MZ, Rajan B, Ostrowski ML, Ramzy I, Schwartz MR, Localized thymic Langerhans’ cell histiocytosis and its relationship with myasthenia gravis: immunohistochemical, ultrastructural, and cytometric studiesArch Pathol Lab Med 1997 121:134-38. [Google Scholar]
[6]. Neumann MP, Frizzera G, The coexistence of Langerhans’ cell granulomatosis and malignant lymphoma may take different forms: report of seven cases with a review of the literatureHum Pathol 1986 17:1060-65. [Google Scholar]
[7]. Egeler RM, Neglia JP, Puccetti DM, Brennan CA, Nesbit ME, Association of Langerhans cell histiocytosis with malignant neoplasmCancer 1993 71:865-73. [Google Scholar]
[8]. Bain BJ, Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB or FGFR1Haematologica 2010 95(5):696-98. [Google Scholar]
[9]. Inhorn RC, Aster JC, Roach SA, Slapak CA, Soiffer R, Tantravahi R, A syndrome of lymphoblastic lymphoma, eosinophilia, and myeloid hyperplasia/malignancy associated with t(8; 13)(pll; qll): description of a distinctive clinicopathologic entityBlood 1995 85(7):1881-18. [Google Scholar]
[10]. Chen W, Wang J, Wang E, Detection of clonal lymphoid receptor gene rearrangements in langerhans cell histiocytosisAm J Surg Pathol 2010 34:1049 [Google Scholar]
[11]. Ohnishi H, Kandabashi K, Maeda Y, Kawamura M, Watanabe T, Chronic eosinophilic leukaemia with FIP1L1–PDGFRA fusion and T674I mutation that evolved from Langerhans cell histiocytosis with eosinophilia after chemotherapyBritish Journal of Haematology 2006 134(5):547-49. [Google Scholar]
[12]. Windebank KP, Nanduri V, Langerhans cell histiocytosisArch Dis Child 2009 94(11):904-08. [Google Scholar]