A Rare Case of Solitary Peutz Jeghers Type Hamartomatous Duodenal Polyp with Dysplasia!
Chetan Devendra Rathi1, Dattatray Balasaheb Solanke2, Nikita Lalitkumar Kabra3, Meghraj Ananda Ingle4, Prabha Dilip Sawant5
1 Senior Resident, Department of Gastroenterology, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai, India.
2 Senior Resident, Department of Gastroenterology, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai, India.
3 Senior Resident, Department of Radiodiagnosis, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai, India.
4 Associate Professor, Department of Gastroenterology, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai, India.
5 Professor and Head, Department of Gastroenterology, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Chetan Devendra Rathi, Senior Resident, Department of Gastroenterology, Lokmanya Tilak Municipal Medical College and General Hospital, Sion, Mumbai – 400022, India.
E-mail: cdrathi@gmail.com
Solitary Peutz Jeghers (SPJ) type hamartomatous polyp is a rare and separate entity from classic Peutz Jeghers syndrome (PJS). A hamartomatous polyp without associated mucocutaneous pigmentations, any other gastrointestinal polyp or a family history of PJS is diagnosed as a SPJ type polyp. We described a case of 22-year-old young man in whom solitary duodenal polyp was incidentally detected and resected. Histopathological examination revealed PJ type hamartomatous polyp with foci of adenomatous and moderate dysplastic change. Very few cases of solitary duodenal PJ type hamartomatous polyps with malignant transformation have been reported. They can be treated with endoscopic or surgical resection. Endoscopic ultrasound can give an idea about the depth of involvement. This is a rare case of duodenal solitary PJ polyp with dysplasia in a young patient.
Contrast enhanced computed tomography, Endoscopic resection, Endoscopic ultrasound, Mucocutaneous pigmentation, Peutz jeghers syndrome
Case Report
A 22-year-old young male came to emergency department with history of blunt abdominal trauma. On ultrasound, there was minimal free fluid in pelvis and so was advised Contrast Enhanced Computed Tomography (CECT) for persistent pain in abdomen.
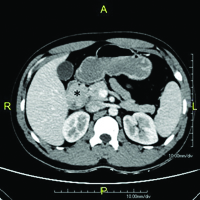
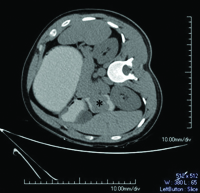
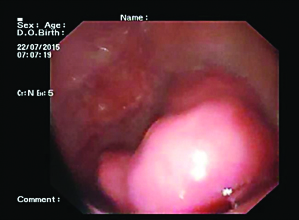
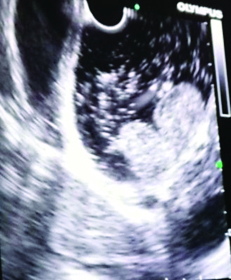
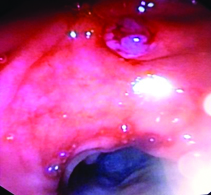
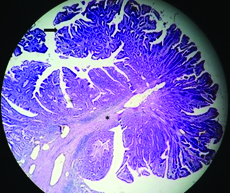
On CECT abdomen and pelvis, there was no solid organ injury or free fluid in abdomen and pelvis. However, there was well defined, lobulated, polypoidal soft tissue density, homogenously enhancing intraluminal mass lesion measuring 4.5x3.8x3.2cm in second part of duodenum occluding the entire lumen [Table/Fig-1,2]. An upper gastrointestinal endoscopy was performed which revealed 4x4cm sessile lobulated polyp in the second part of duodenum with superficial ulcerations and occluding the entire duodenal lumen [Table/Fig-3]. Histological examination of a biopsy specimen obtained from the lesion revealed findings suggestive of a tubulo-villous adenoma with low grade dysplasia. In order to know the exact depth of the polyp, endoscopic ultrasound was done which revealed approximately 5x5cm sized echogenic polypoidal mass in the second part of duodenum with extension of base upto submucosa. Muscularis propria was not involved [Table/Fig-4]. Endoscopic resection was performed [Table/Fig-5] and sample sent for histopathological evaluation revealed Peutz Jeghers type hamartomatous polyp with muscularis mucosa in its arborizing branches with foci of adenomatous changes and moderate dysplasia [Table/Fig-6]. He had no mucocutaneous pigmentation or family history of Peutz Jeghers syndrome. There was no evidence of any other gastrointestinal polyp confirmed on colonoscopy and enterography. Patient is kept on yearly follow for any recurrence or development of new gastrointestinal polyp.
Contrast enhanced CT of abdomen (axial section) demonstrating well-defined soft tissue density homogenously enhancing polypoidal mass in second part of duodenum (asterisk).
Non contrast CT of abdomen (right lateral decubitus view) with positive oral contrast demonstrating well-defined soft tissue density polypoidal mass in second part of duodenum (asterisk).
Upper gastrointestinal endoscopy showing large lobulated sessile polypoidal mass lesion in second part of duodenum.
Endoscopic ultrasound image showing large echogenic mass lesion in second part of duodenum with base extending up to submucosa. Muscularis propria is uninvolved.
Upper gastrointestinal endoscopy after polypectomy.
Histopathology of the resected specimen of duodenal polyp demonstrating branching bundles of smooth muscle fibres (asterisk) covered with duodenal mucosa with a focus of dysplasia (arrow) [Haematoxylin-eosin stain, Magnification x 40].
Discussion
Peutz-Jeghers Syndrome (PJS), first described by Peutz in 1921, is a rare autosomal dominant syndrome caused by germline mutation of serine threonine kinase 11. It is characterized by gastrointestinal hamartomatous polyps and mucocutaneous pigmentation [1]. A hamartomatous polyp without associated mucocutaneous pigmentation or a family history of PJS is diagnosed as a solitary Peutz-Jeghers (PJ) type hamartomatous polyp. Solitary hamartomatous polyp has been considered a variant or a separate disease entity [2]. As compared with PJS, Peutz-Jeghers type hamartomatous polyps are diagnosed with a lower risk of cancer [3].
Patient with PJS are at increased risk for common and unusual types of gastrointestinal and nongastrointestinal tumours. Peutz Jeghers (PJ) type of polyp is defined as a hamartomatous polyp without any clinical symptoms of PJS [4]. It is characterized histologically by tree-like branching of smooth muscle fibres, with a core of smooth muscle covered by mucosal tissue of near normal appearance [2]. A variety of lesions can be found in the duodenum and these lesions can be distinguished based on endoscopic appearance, Endoscopic Ultrasound (EUS) and histology. Biopsy is recommended to determine histology and thus guide management [5]. Solitary PJ type polyps have been described as having a lobular or nodular surface, whitish colour and whitish spots on the surface. It has been suggested that this condition should be considered as a clinical entity different from PJS. Hamartomatous polyps are generally considered to have very low malignant potential [2]. However, some reports have described areas of neoplastic change, such as adenomatous or carcinomatous change in 3-6% of the polyps in PJS [6].
A search of case reports on the MEDLINE database upto July 2015 using the term “solitary duodenal hamartomatous polyp” revealed only 19 well documented cases of solitary duodenal hamartomatous polyp, excluding our case, in 14 studies published in English. Age of onset ranged from 22-89 years (Mean ± SD 58.4±22.31). Only four patients out of 20 were in third decade including our case. Out of 20 cases, 11 were pedunculated, three semi-pedunculated, two sessile including our case and three not specified. Most of the patients underwent endoscopic resection (17 out of 20). Only three patients with polyp size > 50mm required surgical resection. In our case as the polyp was extending upto submucosa with muscularis propria being uninvolved, endoscopic resection was done. Out of 20 patients, five patients showed adenomatous or malignant transformation including our case. Out of five cases with malignant transformation, ours is the youngest patient with dysplasia and sessile polyp, rest four was beyond fourth decade with pedunculated polyp [2]. Because of the malignant potential, a solitary PJ hamartomatous polyp of the duodenum should be removed. There is no definitive guideline for treatment of solitary duodenal PJ type hamartomatous polyp [5]. In our case, patient was young without any other medical illness and the polyp was superficial so endoscopic resection was done.
Conclusion
Solitary duodenal Peutz Jeghers type hamartomatous polyp is a rare entity with low malignant potential. Patient should undergo upper gastrointestinal endoscopy, small bowel enterography, colonoscopy, family screening and look for mucocutaneous pigmentation to differentiate it from Peutz Jeghers syndrome. Endoscopic ultrasonography is useful to determine the depth of lesion to plan treatment. These polyps are treated with endoscopy or surgery depending on size, depth of lesion and evidence of malignant transformation on biopsy.
Consent
Written informed consent was obtained from the patient for publication of this case report.
[1]. Sekino Y, Inamori M, Hirai M, Solitary Peutz-Jeghers type hamartomatous polyps in the duodenum are not always associated with a low risk of cancer: two case reportsJ Med Case Rep 2011 5:240 [Google Scholar]
[2]. Suzuki K, Higuchi H, Shimizu S, Nakano M, Serizawa H, Morinaga S, Endoscopic snare papillectomy for a solitary Peutz-Jeghers type polyp in the duodenum with ingrowth into the common bile duct: Case reportWorld J Gastroenterol 2015 21:8215-20. [Google Scholar]
[3]. Kitaoka F, Shiogama T, Mizutani A, A solitary Peutz-Jeghers-type hamartomatous polyp in the duodenum. A case report including results of mutation analysisDigestion 2004 69:79-82. [Google Scholar]
[4]. Giardiello FM, Trimbath JD, Peutz-Jeghers syndrome and management recommendationsClin Gastroenterol Hepatol 2006 4:408-15. [Google Scholar]
[5]. Basford PJ, Bhandari P, Endoscopic management of nonampullary duodenal polypsTherap Adv Gastroenterol 2012 5:127-38. [Google Scholar]
[6]. Suzuki S, Hirasaki S, Ikeda F, Yumoto E, Yamane H, Matsubara M, Three cases ofSolitary Peutz-Jeghers-type hamartomatous polyp in the duodenumWorld J Gastroenterol 2008 14(6):944-47. [Google Scholar]