Benign Recurrent Intrahepatic Cholestasis in a Young Adult
Prabhat Kumar1, Riyaz Charaniya2, Arvind Ahuja3, Sakshi Mittal4, Ratnakar Sahoo5
1 Senior Resident, Department of Medicine, PGIMER & Dr RML Hospital, New Delhi, India.
2 Postgraduate Resident, Department of Medicine, PGIMER & Dr RML Hospital, New Delhi, India.
3 Associate Professor, Department of Pathology, PGIMER & Dr RML Hospital, New Delhi, India.
4 Senior Resident, Department of Medicine, PGIMER & Dr RML Hospital, New Delhi, India.
5 Professor, Department of Medicine, PGIMER & Dr RML Hospital, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Prabhat Kumar, Senior Resident, Department of Medicine, OPD Block, PGIMER & Dr RML Hospital, Baba Kharak Singh Marg, New Delhi-110001, India.
E-mail: drkumar.prabhat@gmail.com
Benign Recurrent Intrahepatic Cholestasis (BRIC) is a rare genetic disorder characterized by recurrent episodes of cholestatic jaundice. The initial episode of jaundice generally occurs before second decade of life and can persist for several weeks to months before resolving spontaneously. It is a benign disease and even after repeated episodes of jaundice, fibrosis of liver cell does not occur. We had a young adult patient who was having recurrent episodes of cholestatic jaundice with intervening symptom free period for last 20 years. He had first episode of jaundice at the age of eight and since then had several similar episodes. Diagnosis was made by classical clinical presentation and histopathological findings. We intend to report this case due to rarity of this disease in India.
Cholestatic jaundice, Pruritus, Rifampicin
Case Report
A 27-year-old male presented with complaints of jaundice for two weeks which was associated with clay coloured stool, dark coloured urine and pruritus. There was no history of pain abdomen, drug exposure, fever, rash, blood transfusion or alcohol intake. He had several similar episodes of jaundice in the past. First episode of jaundice and pruritus occurred at the age of eight years which resolved spontaneously within a month and second episode occurred four years later, which also resolved in six weeks. Since then patient reported to have repeated episodes of jaundice and pruritus once in every two-three years. Each episode lasted for about a month or two, with symptom free interval in between. He used to visit a local doctor in his town during his illness where he was given anti histaminics and ursodeoxycholic acid for his complaints. He was never evaluated thoroughly for these symptoms and was never advised to visit a higher centre. There were no similar complaints in any of his family members. On physical examination, he had deep icterus with no signs of liver cell failure. Liver was just palpable below the right costal margin. A few scratch marks were noted over his body.
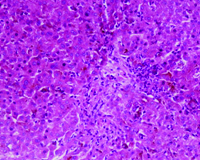
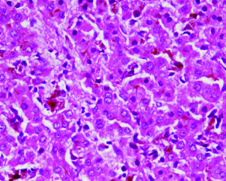
Investigations showed haemoglobin of 13.5 g/dl, Total leukocyte count of 10,000/cumm and platelet count of 2.9 lac/cumm. Liver function tests were deranged. Total Bilirubin was 24 mg/dl of which conjugated fraction was 20.1 mg/dl, Aspartate aminotranferase (AST) and Alanine aminotransferase (ALT) was 94 and 99 IU/L respectively (normal<50 IU/L), Alkaline phosphatase was 421 U/L (normal: 30-120 U/L), Gamma glutamyltransferase (GGT) was 56 U/L (normal: 8-61 U/L), prothrombin time was 16 seconds (control 15 seconds). Other biochemical parameters including renal function tests and serum electrolytes were normal. Hepatitis B surface antigen (HBsAg), antibodies to hepatitis C virus (Anti HCV), IgM antibodies to hepatitis A virus (IgM anti HAV) and IgM antibodies to hepatitis E virus (IgM anti HEV) were negative. Antinuclear antibodies (ANA), antimitochondrial antibodies, anti smooth muscle antibodies, anti liver kidney microsomal antibodies and serum ceruloplasmin levels were also normal. Ultrasonography of abdomen revealed mild hepatomegaly with normal echotexture. Magnetic resonance cholangiopancreatography (MRCP) showed normal intra and extra hepatic biliary tree and pancreatic ductal system. Liver biopsy showed intrahepatic and canalicular cholestasis predominantly involving zone 3 [Table/Fig-1] with pigment laden Kupffer cells [Table/Fig-2]. These features were suggestive of Benign Recurrent Intrahepatic Cholestasis (BRIC).
Low power photomicrograph showing intrahepatic as well as canalicular cholestasis in zone 3 (H&E x100).
High power photomicrograph to better appreciate the cholestasis with sinusoids showing pigment laden kupffer cells (H&E x400).
He was treated with Ursodeoxycholic acid 300 mg thrice daily and cholestyramine 4 grams twice daily. He was advised to take low fat diet and fat soluble vitamins were supplemented. After a month his bilirubin was 5 mg/dl which came to baseline after two months of therapy. Patient was followed up for six months and has not developed fresh episode of pruritus and jaundice.
Discussion
Benign Recurrent Intrahepatic Cholestasis (BRIC) is a rare genetic disorder characterized by intermittent episodes of jaundice and pruritus. It is a benign disease with no progression to end stage liver disease [1]. First case of BRIC was described by Summerskill and Walshe in 1956 and hence also known as Summerskill-Walshe-Tygstrup syndrome.
Age at first presentation can range from 1 to 59 years and each episode can last for several weeks to months. The number of attacks can be one in a decade or several in a year. There is completely asymptomatic phase in between attacks which can last from months to years [2]. Cholestatic episode generally begins before second decade of life and prodromal symptoms include nausea, vomiting, pruritus, malaise and weight loss. It is followed by an icteric phase which is characterized by conjugated hyperbilirubinaemia and raised alkaline phosphatase levels. Serum gamma-glutamyl transpeptidase (GGT), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels are either normal or mildly raised. Each episode last for weeks to months and is followed by complete clinical and biochemical resolution. During recovery, pruritus is first to resolve completely followed by normalization of deranged bilirubin and transaminase levels.
Liver biopsy is characterized by noninflammatory intrahepatic cholestasis and Kupffer cell hyperplasia. There is no progression to cirrhosis and liver histology remains normal during remission [3]. A diagnostic criteria for BRIC has been proposed by Luketic and Shiffman which includes: (a) At least two episodes of jaundice with asymptomatic interval of months to years; (b) Laboratory investigations suggestive of intrahepatic cholestasis; (c) Cholestasis induced severe pruritus; (d) Cholangiography showing normal intra and extrahepatic bile ducts; (e) Liver histology suggesting centrilobular cholestasis; (f) Absence of other causes of cholestasis [4]. Our patient satisfied all these criteria and was having symptomatic obstructive jaundice since age of eight years with completely symptom free intervals.
BRIC is an autosomal recessive disorder and is associated with mutation of ATP8B1 and ABCB11 genes. BRIC has been further subclassified into two types. BRIC-1 and BRIC-2 are caused by mutation of ATP8B1 and ABCB11 gene respectively [5]. Defective ATP8B1 gene is located on chromosome 18 and encodes F1C1 which causes impaired bile salt excretion while defective ABCB11 gene is located on chromosome 2 which causes impaired functioning of canalicular pump leading to cholestasis. Both subtypes have clinical differences like pancreatitis is seen in BRIC-1 and cholelithiasis has been described in BRIC-2 [6].
There is no definite treatment for BRIC and medications are used during an episode to alleviate the symptoms. Drugs like bile acid sequestrants, centrally acting opiod antagonists, antihistaminics and rifampicin are used for pruritus. It is believed that bile acid retention within body causes pruritus in these patients. Cholestyramine causes loss of bile acid by binding with them, which are excreted in the feces [7]. Rifampicin decreases duration of cholestasis by inhibiting uptake of bile acids by hepatocytes. Short term nasobiliary drainage improves pruritus in cholestatic jaundice [8]. Extracorporeal albumin dialysis in a Molecular Absorbent Recycling System (MARS) removes bile acids from body and causes rapid improvement in pruritus [9]. Partial biliary diversion is also effective in decreasing pruritus by reducing bile acid load on the ileal transport system [10]. Liver transplantation is rarely done for intractable pruritus [11]. Our patient improved with cholestyramine and ursodeoxycholic acid therapy.
Conclusion
Benign recurrent intrahepatic cholestasis is a self limiting disease and does not cause chronic liver disease. Knowledge of this entity is crucial as an early recognition can prevent performance of expensive diagnostic investigations and patient can be counseled regarding benign nature of this disease.
[1]. Parolin MB, Langowiski AR, Ioshii SO, Maggio EM, Coelho JC, Benign recurrent intrahepatic cholestasis: a seven-year follow-up reportArq Gastroenterol 2000 37:231-34. [Google Scholar]
[2]. Luketic VA, Shiffman ML, Benign recurrent intrahepatic cholestasisClin Liver Dis 2004 8:133-49. [Google Scholar]
[3]. Biempica L, Gutstein S, Arias IM, Morphological and biochemical studies of benign recurrent cholestasisGastroenterology 1967 52(3):521 [Google Scholar]
[4]. Luketic VA, Shiffman ML, Benign recurrent intrahepatic cholestasisClin Liver Dis 1999 3:509-28. [Google Scholar]
[5]. Van Mil SW, Van der Woerd WL, Van der Brugge G, Sturm E, Jansen PL, Bull LN, Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11Gastroenterology 2004 127(2):379-84. [Google Scholar]
[6]. Van der Woerd WL, Van Mil SW, Stapelbroek JM, Klomp LW, van de Graaf SF, Houwen RH, Familial cholestasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancyBest Pract Res Clin Gastroenterol 2010 24(5):541 [Google Scholar]
[7]. Spiegel EL, Schubert W, Perrin E, Schiff L, Benign recurrent intrahepatic cholestasis with response to cholestyramineAm J Med 1965 39(4):682-88. [Google Scholar]
[8]. Stapelbroek JM, van Erpecum KJ, Klomp LW, Venneman NG, Schwartz TP, van Berge Henegouwen GP, Nasobiliary drainage induces long-lasting remission in benign recurrent intrahepatic cholestasisHepatology 2006 43:51-53. [Google Scholar]
[9]. Stange J, Hassanein TI, Mehta R, Mitzner SR, Bartlett RH, The molecular adsorbents recycling system as a liver support system based on albumin dialysis: A summary of preclinical investigations, prospective, randomized, controlled clinical trial, and clinical experience from 19 centersArtif Organs 2002 26:103-10. [Google Scholar]
[10]. Hollands CM, Rivera-Pedrogo FJ, Gonzalez-Vallina R, Loret-de-Mola O, Nahmad M, Burnweit CA, Ileal exclusion for Byler’s disease: an alternative surgical approach with promising early results for pruritusJ Pediatr Surg 1998 33:220-24. [Google Scholar]
[11]. Mezey E, Burns C, Burdick JF, Braine HG, A case of severe benign intrahepatic cholestasis treated with liver transplantationAm J Gastroenterol 2002 97:475-77. [Google Scholar]