A Case Report of Childhood Recurrent Autoimmune Pancreatitis: A Rare Emerging Entity
Mohammed Y Hasosah1, Lojain Masawa2, Ajwan Jan3, Khalid Alsaleem4
1 Assistant Professor, Department of Pediatric Gastroenterology, King Saud Bin Abdulaziz University for Health Sciences, National Guard Hospital, Jeddah, Saudi Arabia.
2 Paediatric Intern, Department of Pediatrics, King Saud Bin Abdulaziz University for Health Sciences, National Guard Hospital, Jeddah, Saudi Arabia.
3 Paediatric Intern, Department of Pediatrics, King Saud Bin Abdulaziz University for Health Sciences, National Guard Hospital, Jeddah, Saudi Arabia.
4 Consultant Gastroenterologist, Department of Pediatrics, Division of Gastroenterology, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Mohammed Hasosah, Assistant Professor of Pediatric, Department of Pediatric Gastroenterology, King Saud Bin Abdulaziz University for Health Sciences, Pediatric Consultant Gastroenterologist, King Abdul-Aziz Medical City, National Guard Hospital, Jeddah, PO Box: 8202, Jeddah-21482, Saudi Arabia.
E-mail: hasosah2007@yahoo.com
Autoimmune pancreatitis (AIP) is a rare entity in the paediatric population and its aetiology is unknown. Here, we report a 10-year-old girl with recurrent abdominal pain. A diagnosis of AIP was made based on elevated pancreatic enzymes, elevated IgG 4 and image findings. She responded to corticosteroid treatment. AIP should be considered in the differential diagnosis of recurrent pancreatitis. Correct diagnosis can help avert the consequences of progressive disease and unnecessary surgery.
Corticosteroid, Pancreatic enzymes, Recurrent abdominal pain
Case Report
A 10-year-old girl was referred to our institution for evaluation of recurrent abdominal pain. She has been suffering recurrent severe right upper quadrant pain and vomiting for three years which resolved following conservative treatment. The parents reported that her appearance was normal during the first seven year of her life. Subsequently, they had noted that she appeared thinner and fatigue. There was no consanguinity and no family history of autoimmune disorders. Her physical examination revealed no pallor or jaundice. Her heart rate was 98/min., blood pressure 100/60 mm/hg and body temperature 36.70C. Abdominal examination revealed mild tenderness but no hepatosplenomegaly. Her laboratory results were as following; haemoglobin: 11.9 (normal range: 12.2-15.3 gm/dL), White Blood Cell: 6.8 (normal range: 6-16×109/L), Platelet: 172 (normal range: 150-450×109/L), serum triglycerides: 420 (normal range: < 150 mg/dl), calcium 9.9 (normal range: 8 to 10.5 mg/dL), serum amylase: 1560 (normal range: 40-140 U/L) and serum lipase: 3230 (normal range: 0-160 U/L). Serum alanine transaminase, aspartate aminotransferase, alkaline phosphatase, gammaglutamyl transferase, albumin and bilirubin were normal. Viral screens (Hepatitis A Virus, Hepatitis B Virus, Hepatitis C Virus, Epstein-Barr virus, Cytomegalovirus and Herpes Simplex Virus) were negative.
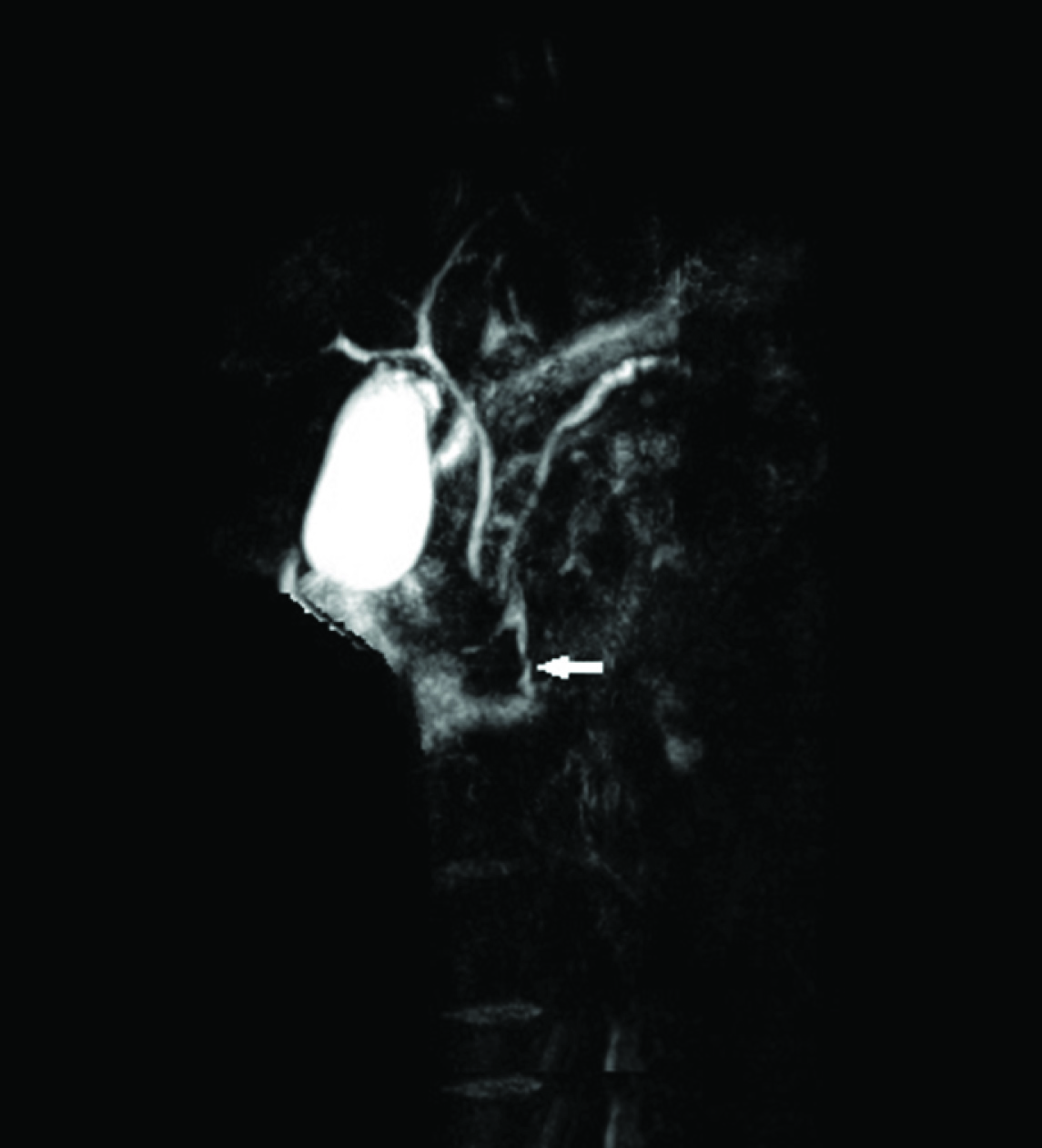
The child was admitted as recurrent pancreatitis. The differential diagnosis included hereditary pancreatitis, structural pancreatitis, inflammatory pseudo tumour and atypical presentation of cystic fibrosis. Abdominal ultrasound showed an enlarged pancreatic head (20×15 mm). The parenchyma was slightly heterogeneous. No stones in biliary or pancreatic ducts. Magnetic resonance cholangiopancreatography (MRCP) showed diffusely enlarged pancreas with fat surrounding its tail and the irregular main pancreatic duct [Table/Fig-1]. No pancreas divisum or stones. Screen for hereditary pancreatitis mutations includes the following; SPINK-1 gene: serine protease inhibitor, Kazal type-1 and PRSS1 gene (protease, serine, 1 (trypsin 1) were negatives. Screens for autoantibodies (anti-nuclear antibodies, anti-DNA antibodies, anti–smooth muscle antibodies, rheumatoid factor, anti-neutrophil cytoplasmic antibodies) were negative. Anti-liver/kidney microsome (LKM) was positive. Serum Immunoglobulin G (IgG) was elevated at 21.7 (normal range: 4.9-16.1 g/L). Serum IgG4 level: 210 (normal range : < 140 mg/dl). A diagnosis of autoimmune pancreatitis was made based on elevated pancreatic enzymes, elevated IgG 4 and image findings. The patient was treated with oral prednisolone 1 mg/kg/day. At 6 months follow-up from the onset of treatment, she was completely asymptomatic, pancreatic duct was reduced, and her pancreatic enzymes remained normal.
Magnetic Resonance Cholangiopancreatography (MRCP) showed diffused swelling of pancreas with low attenuation surrounding pancreas and narrowing of the main pancreatic duct (arrows)
Discussion
Autoimmune pancreatitis (AIP) is a chronic disease of unknown cause and is characterized by raised levels of serum IgG4 and responsiveness to immunosuppressive therapy [1]. AIP is a rare cause of recurrent pancreatitis in the adult and paediatric populations. Accurate diagnosis of AIP relied on characteristic clinical, histologic and morphologic findings. However, autoimmune disease was presumed an aetiology of AIP [2]. The first reported of AIP was made in 1959, when defined AIP as a consequence of hypergammaglobulinaemia [3]. Only 16 children with AIP have been reported in the literature [4]. Zen et al., described the predominant type of AIP in children appears to be type 2, given that more than a half of reported paediatric cases had granulocytic epithelial lesions (GELs), whereas “definitive type 1 AIP” has never been described in children [5]. Diagnosis of AIP is based primarily on the following criteria: 1) diffuse enlargement of the pancreas and irregular narrowing of the main pancreatic duct; 2) presence of autoantibodies; 3) increased serum gammaglobulin or IgG levels; 4) cholestatic liver dysfunction; 5) fibrosis with lymphocytic infiltration in the pancreas; and 6) responsiveness to steroid therapy [6]. In our patient, based upon the combination of recurrent symptoms, presence of autoantibodies, MRI findings and elevated IgG4 level: 210 (normal range : <140 mg/dl), a diagnosis of AIP was made. AIP should be considered in any autoimmune conditions which involved the pancreas and biliary tract. Correct diagnosis can help avert the consequences of progressive disease and unnecessary surgery. Histological diagnosis of AIP in children is difficult because tissue acquisition can be difficult to obtain by endoscopic ultrasound with Fine-Needle Aspiration (FNA).
The pathogenesis of AIP is unknown. However, there is a linkage between AIP and IgG4-positive plasma cells. The usefulness of serum IgG4 in diagnosing AIP had been evaluated in a meta-analysis of seven studies. Serum IgG4 showed sensitivity (67–94%) and specificity (89–100%) in diagnosing AIP [7]. Antibodies to carbonic anhydrase II antigens, rheumatoid factor, antinuclear antibody and anti-smooth muscle antibody are other antibodies associated with AIP but they are not diagnostic for AIP [8].
The treatment of AIP is based mainly on steroid therapy [9]. Steroid therapy is appropriate only in symptomatic cases, allowing improvement of the clinical symptoms in a few days and the biological disorders in the first 2 weeks of treatment, followed by improvement of the morphological anomalies affecting the pancreas and its ducts [9]. In the present case, steroid therapy allowed clinical and radiological improvement.
Conclusion
This case is reported for its rare and challenging diagnostic presentation. The early identification of child with AIP as potential cause of recurrent pancreatitis is important, because delayed treatment of AIP might lead to irreversible consequences and avoid morbidity and mortality
[1]. Okazaki K, Uchida K, Koyabu M, Miyoshi H, Takaoka M, Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related diseaseJournal of Gastroenterology 2011 46(3):277-88. [Google Scholar]
[2]. Finkelberg DL, Sahani D, Deshpande V, Brugge WR, Autoimmune pancreatitisN Engl J Med 2006 355:2670 [Google Scholar]
[3]. Sarles H, Sarles JC, Muratore R, Chronic inflammatory sclerosis of the pancreas: an autonomous pancreatic disease?Am J Dig Dis 1961 6:688-98. [Google Scholar]
[4]. Fujii LL, Chari ST, El-Youssef M, Paediatric pancreatic EUS-guided trucut biopsy for evaluation of autoimmune pancreatitisGastrointest Endosc 2013 77:824-28. [Google Scholar]
[5]. Zen Y, Grammatikopoulos T, Hadzic N, Autoimmune pancreatitis in children: insights into the diagnostic challengeJ Paediatr Gastroenterol Nutr 2014 59(5):e42-45. [Google Scholar]
[6]. Yoshida K, Toki F, Takeuchi T, Chronic pancreatitis caused by autoimmune abnormality: proposal of concept of autoimmune pancreatitisDig Dis Sci 1995 40:1561-68. [Google Scholar]
[7]. Morselli-Labate AM, Pezzilli R, Usefulness of serum IgG4 in the diagnosis and follow up of autoimmune pancreatitis: A systematic literature review and meta-analysisJ Gastroenterol Hepatol 2009 24:15-36. [Google Scholar]
[8]. Kim KP, Kim MH, Song MH, Autoimmune chronic pancreatitisAm J Gastroenterol 2004 99:1605-16. [Google Scholar]
[9]. Park DH, Kim MH, Chari ST, Recent advances in autoimmune pancreatitisGut 2009 58:1680-89. [Google Scholar]