β-Thalassaemia and its Co-existence with Haemoglobin E and Haemoglobin S in Upper Assam Region of North Eastern India: A Hospital Based Study
Anju Barhai Teli1, Rumi Deori2, Sidhartha Protim Saikia3, Kalyani Pathak4, Rita Panyang5, Rashmi Rajkakati6
1 Associate Professor, Department of Biochemistry, Assam Medical College & Hospital, Dibrugarh, Assam, India.
2 Assistant Professor, Department of Biochemistry, Assam Medical College & Hospital, Dibrugarh, Assam, India.
3 Senior Research Fellow, Department of Anatomy, Assam Medical College & Hospital, Dibrugarh, Assam, India.
4 Junior Research Fellow, Department of Biochemistry, Assam Medical College & Hospital, Dibrugarh, Assam, India.
5 Assistant Professor, Department of Paediatrics, Assam Medical College & Hospital, Dibrugarh, Assam, India.
6 Associate Professor, Department of Biochemistry, Assam Medical College & Hospital, Dibrugarh, Assam, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Anju Barhai Teli, Associate Professor, Department of Biochemistry, Assam Medical College & Hospital, Dibrugarh-786002, Assam, India.
E-mail: dr.anjub.t@gmail.com
Introduction
β-Thalassaemias are common genetic disorders in the Indian subcontinent and its status has not been well studied in the Upper Assam region of North Eastern India.
Aim
The aim of the study was to show the prevalence of β- thalassaemias and its co-existence with Haemoglobin E and Haemoglobin S in the Upper Assam region of North Eastern India.
Materials and Methods
A total of 1200 anaemic patients were investigated for β- thalassaemias. Complete Blood Count (CBC) and High Performance Liquid Chromatography (HPLC) were done for screening.
Results
Out of 1200 patients screened, 5.83% β-thalassaemia trait, 2.33% compound Hb E/β-Thalassaemia, 1.33% β-thalassaemia major and 0.42% compound Hb S/β- thalassaemia were detected. A high incidence of thalassaemia is found among the people of Upper Assam region of North Eastern India.
Conclusion
The only way to prevent the disease is carrier detection and awareness among the people about it.
Anaemia, Complete blood count, HPLC
Introduction
β- Thalassaemia is a monogenic single gene disorder characterized by defective synthesis of the β-globin chain. In India β-thalassaemia is prevalent in almost every population; however the prevalence is high among the Sindhis, Gujaratis, Bengalis, Punjabis and Muslims [1].
Among the common Hb variants, Hb E is commonly found in the North-Eastern states of India and the average allele frequency of Hb E in North-East region is 10.9% [2]. Co-existence of β-thalassaemia with Hb E develops a condition which is very much similar to thalassaemia major and mild form of thalassaemia intermedia [3]. Interaction of β-thalassaemia with Hb E and Hb S is becoming a major health problem in India [4]. It should, however, be noted that the calculated numbers of β-thalassaemia heterozygotes in India assumed a carrier frequency of 3–4% [5].
Aim
This study was planned to show the status of β-thalassaemia along with its co-existence with Hb E and Hb S in the Upper Assam region of North East India.
Materials and Methods
The present study was carried out among the patients sent to the Healthcare Clinical Biochemistry Laboratory of Assam Medical College & Hospital, Dibrugarh, Assam, India by clinicians as a work up for anaemia and for confirmation of β-thalassaemia. All the patients were from different districts of Upper Assam region of North East India [Table/Fig-1]. Due clearance was obtained from Institutional ethics committee for this study. This study was carried out from January 2012 to June 2015.
Map of Assam showing the study sites.

A total of 1200 patients who were between 1-65 years of age were studied. Detail clinical histories including ethnic origin, age, sex, blood transfusion etc. along with family history were recorded.
4 ml of venous blood was collected in EDTA vials after obtaining informed consent from each individual. Complete Blood Count (CBC) was done in cell counter (SYSMEX XS- 800i, Japan) using standard procedure. The screening of β- thalassaemia was done by a High-Performance Liquid Chromatography (HPLC) – based D10 Haemoglobin Testing System (BioRad Laboratories, USA). HPLC is one of the best methods used for accurate detection of various haemoglobin variants and β-thalassaemia trait [6,7]. HbA2 level of >4.0% was used as a cut-off for diagnosis of β-thalassaemia trait. After analysis of the samples, parents and siblings of patients found to have β-thalassaemia trait, compound Hb E/β-thalassaemia, β-thalassaemia major and compound Hb S/β-thalassaemia were called for investigation for β-thalassaemia and follow-up for counseling if required.
Statistical Analysis
Statistical analysis was done by using SPSS 21. The Mean and SD were calculated by this software.
Results
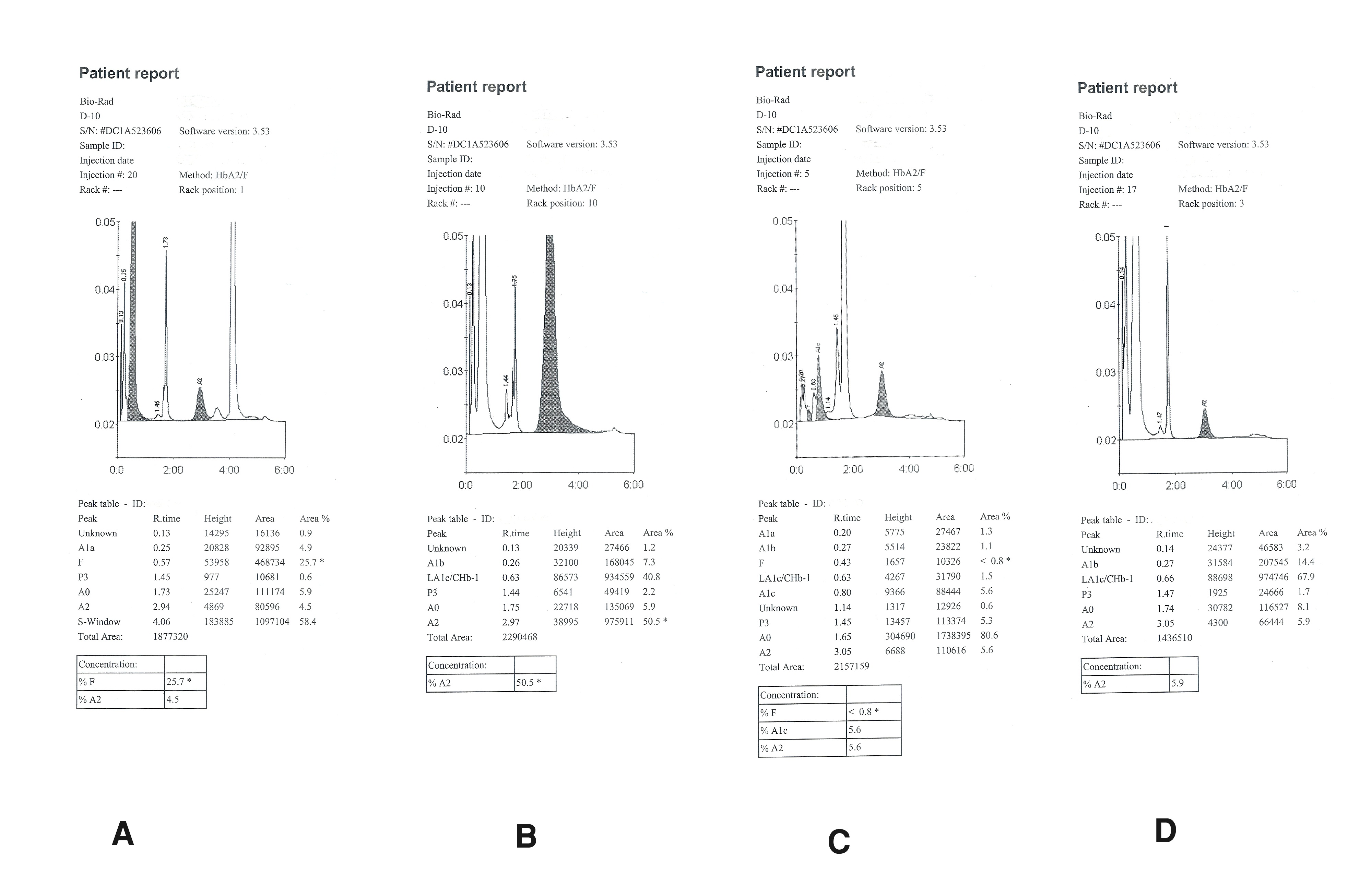
Among the 1200 studied patients males had a mean Hb of 8.36 ± 3.46 g/dl while females had a mean Hb of 7.18 ± 3.1 g/dl. In our study 5.83% β-thalassaemia trait (70 patients), 2.33% compound Hb E/β-thalassaemia (28 patients), 1.33% β-thalassaemia major (16 patients) and 0.42% compound Hb S/β-thalassaemia (5 patients) were detected. The types of chromatograms of the patients were shown in the [Table/Fig-2]. [Table/Fig-3] shows the haematological data [Mean ± SD] of the patients. Anaemia is the most common sign observed in all these patients. All the clinical signs found among the patients are shown in the [Table/Fig-4]. In this study β-thalassaemia trait is the commonest abnormality encountered. The haematological data shows mean Hb of 9.232 g/dl. The patients of Hb E/β-thalassaemia shows moderate to severe anaemia (mean Hb 5-7g/dl) [8]. In our study the mean Hb concentration among the Hb E/β-thalassaemia patients is 6.362 g/dl. The patients of β-thalassaemia major shows severe anaemia (mean Hb 4.678 g/dl). The patients with Hb S/β-thalassaemia shows mean Hb of 5.4 g/dl.
Chromatograms of the patients of: (a) Hb S/β-thalassaemia; (b) Hb E/β-thalassaemia; (c) β-thalassaemia trait; and (d) β-thalassaemia major detected during the study
Haematological data (Mean ± SD) of the patients of β-thalassaemia trait, β -thalassaemia major, Hb E/β-thalassaemia and Hb S/β- thalassaemia detected during the study.
| Group | RBC(×106/ μl) | Hb (g/dl) | MCV (fl) | MCH (pg) | MCHC (g/dl) |
|---|
| β- thalassaemia trait | 4.468 ± 1.1564 | 9.232± 2.6436 | 71.242± 13.7913 | 21.656±3.3932 | 29.895±2.6782 |
| β-thalassaemia major | 3.342 ± 2.1758 | 4.678± 3.7285 | 69.111±9.3420 | 20.989± 2.5042 | 30.522± 4.1505 |
| Hb E/β-thalassaemia | 2.957 ± 1.2456 | 6.362± 2.7703 | 69.793± 16.2261 | 20.407± 5.2068 | 29.300± 3.2711 |
| Hb S/β- thalassaemia | 2.133 ± 1.2097 | 5.400± 2.6907 | 89.600± 5.1740 | 25.767± 1.6166 | 28.733± 1.2702 |
Clinical signs observed among the patients of β-thalassaemia trait, β -thalassaemia major, Hb E/β-thalassaemia and Hb S/β- thalassaemia.
| Clinical signs | β-thalassaemia trait | Hb E/β-thalassaemia | β-thalassaemia major | HbS/β- thalassaemia |
|---|
| Anaemia | 80% | 42.86% | 100% | 100% |
| Splenomegally | 7.14% | 39.29% | 62.5% | - |
| Hepato-Splenomegally | - | 14.29% | 25% | 40% |
| Weakness | 41.43% | 35.71% | 100% | 80% |
| Fever | 1.43% | - | - | - |
| Stomach Pain | - | 7.14% | - | - |
| Gall stones | 4.29% | 3.57% | - | - |
Discussion
Thalassaemia is an inherited autosomal recessive blood disorder. Previously it was confined to certain areas, religions, castes and tribes but the prevalence has increased globally due to the migration of people from one place to another and inter-community marriages [9].
It is evident from the present findings that β-thalassaemia is in abundance in endemic form in the present population. The prevalence of β-thalassaemia trait and sickle cell in India varies between 3-17% and 1-44% respectively because of consanguinity and caste and area endogamy [10,11]. It is interesting to note that the proliferation of abnormal Hb S and β-thalassaemia in Assam is due to caste and area endogamy. These many aspects are the root causes for increasing complexities of Sickle Cell Disease and β-thalassaemia in Assam. The Hb S/β-thalassaemia patients have more severe disease with lower Hb level, MCV and MCH than their counterparts having Hb SS [12]. The red cell count was relatively higher among Hb S/β-thalassaemia patients in relation of the haemoglobin and MCH in carriers of the thalassaemia may be due to production of extra microcytes.
Globally among the patients with severe β-thalassaemia, about 50% of them are affected by Hb E/β-thalassaemia genotype [13–20]. The prevalence rate of Hb E/β-thalassaemia is highest in India, Bangladesh and Southeast Asia [18,19,21].
In this study, the prevalence of Hb E/β-thalassaemia was 2.33% of the overall study population. The previous reports showed that the prevalence of Hb E/β-thalassaemia in northeast India is 9% [22], in upper Assam region is 1.26% [23] and in Dibrugarh it is 1.44% [24]. In this study all the patients with Hb E/β-thalassaemia were found to be severely anaemic. [Table/Fig-5] shows the comparison of the prevalence data of β-thalassaemia between this study and the other studies from India [22–26]. Patient with Hb E/β-thalassaemia carries one gene for β-thalassaemia from one parent and one gene for Hb E from the other parent and hence shows clinical manifestation similar to β-thalassaemia major or Hb E disease. In our study, the numbers of Hb E/β-thalassaemia patients are more than the number of β-thalassaemia major patients.
Comparison of the prevalence data of β-thalassaemia between this study and the other studies from India.
| Sl. No. | Study Site | Number Screened | β- Thalassaemia Trait (%) | Hb E / β- Thalassaemia (%) | Hb S / β- Thalassaemia (%) | β - Thalassaemia Major (%) | Study |
|---|
| 1 | Upper Assam | 1200 | 5.83 | 2.33 | 0.42 | 1.33 | Present study |
| 2 | Assam/Meghalaya/ Arunachal Pradesh | 800 | 18.12 | 9 | 0.13 | 2.12 | Pathak et al., [22] |
| 3 | Upper Assam | 9000 | 3.48 | 1.26 | 0.59 | 0.36 | Baruah et al., [23] |
| 4 | Bangalore | 9981 | 2.16 | 0.01 | 0.06 | - | Mohanty et al., [24] |
| 5 | Kolkata | 9990 | 3.64 | 0.09 | - | - | Mohanty et al., [24] |
| 6 | Dibrugarh | 6816 | 1.48 | 1.44 | 0.04 | - | Mohanty et al., [24] |
| 7 | Ludhiana | 9991 | 3.96 | - | - | - | Mohanty et al., [24] |
| 8 | Mumbai | 10011 | 2.55 | - | - | - | Mohanty et al., [24] |
| 9 | Vadodara | 9991 | 2.68 | 0.01 | - | - | Mohanty et al., [24] |
| 10 | South India | 543 | 37.9 | 4.6 | 2.5 | 2.3 | Chandrashekar [26] |
To estimate the disease burden of β-thalassaemias and other haemoglobin variants micromapping is important because the frequency of the disease varies widely even within small geographic regions in different countries [25].
Limitations
Hb electrophoresis is not performed in this study which is another method for detection of β-thalassaemia and other Hb variants.
Conclusion
This study provides a comprehensive data on the status of thalassaemia in Upper Assam region of North Eastern India. The study shows that the prevalence of β-thalassaemia is high and it is causing a severe public health problem in this region. Since there is no effective treatment for this disease, so carrier detection and awareness programmes have to be conducted at community level for its control and prevention. Advance molecular techniques like polymerase chain reaction (PCR), direct sequencing should be used followed by genetic counseling of the patients.
[1]. Agarwal MB, Mehta BC, Genotypic analysis of symptomatic thalassaemia syndromes (A study of 292 unrelated cases from Bombay)J Postgrad Med 1982 28:1-3. [Google Scholar]
[2]. Balgir RS, Genetic epidemiology of the three predominant abnormal haemoglobins in IndiaJ Assoc Physicians India 1996 44:25-28. [Google Scholar]
[3]. Galanello R, Origa R, Beta-thalassaemiaOrphanet J Rare Dis 2010 5:11 [Google Scholar]
[4]. Colah RB, Surve R, Sawant P, D’Souza E, Italia K, Phanasgaonkar S, HPLC Studies in HaemoglobinopathiesIndian J Pediatr 2007 74:657-62. [Google Scholar]
[5]. Joint WHO–TIF meeting on management of haemoglobin disorders (2nd: 2008: Nicosia, Cyprus) Geneva, World Health Organization. (NLM classification: WH 190) [Google Scholar]
[6]. Bravo-Urquiola M, Arends A, Montilla S, Velasquez D, Garcha G, Alvarez M, Advantage in the use of high performance chromatography technique for screening haemoglobinopathies in VenezuelaInvest Clin 2004 45:309-15. [Google Scholar]
[7]. Moorchune N, Phillip J, Sarkar RS, Prasad R, Dutta V, Is high pressure liquid chromatography an effective screening tool for characterization of molecular defects in haemoglobinopathies?Indian J Pathol Microbiol 2013 56:36-39. [Google Scholar]
[8]. Vichinsky E, Haemoglobin E syndromesHematology Am Soc Hematol Educ Program 2007 :79-83. [Google Scholar]
[9]. Patne SC, Shukla J, Haemoglobin E disorders in Eastern Uttar PradeshIndian J Pathol Microbiol 2009 52:110-12. [Google Scholar]
[10]. Balgir RS, The Burden of Haemoglobinopathies in India and the Challenges AheadCurrent Science 2000 79:1536-47. [Google Scholar]
[11]. Balgir RS, The Genetic Burden of Haemoglobinopathies with Special Reference to Community Health in India and the Challenges AheadIndian J Hematol Blood Transfus 2002 20:2-7. [Google Scholar]
[12]. Urade BP, Incidence of sickle cell anaemia and thalassaemia in central IndiaOp J Blood Dis 2012 :71-80. [Google Scholar]
[13]. Modell B, Darlison M, Global epidemiology of haemoglobin disorders and derived service indicatorsBull World Health Organ 2008 86:480-87. [Google Scholar]
[14]. Chen S, Eldor A, Barshtein G, Zhang S, Goldfarb A, Rachmilewitz E, Enhanced aggregability of red blood cells of beta-thalassaemia major patientsAm J Physiol 1996 270:1951-56. [Google Scholar]
[15]. de Silva S, Fisher CA, Premawardhena A, Lamabadusuriya SP, Peto TE, Perera G, Thalassaemia in Sri Lanka: implications for the future health burden of Asian populations. Sri Lanka Thalassaemia Study GroupLancet 2000 355:786-91. [Google Scholar]
[16]. Premawardhena A, De Silva S, Arambepola M, Olivieri N, Merson L, Muraco J, Thalassaemia in Sri Lanka: a progress reportHum Mol Genet 2004 13:203-06. [Google Scholar]
[17]. Vichinsky EP, Report of proceedings: 1999 international conference on E-B thalassaemiaJ Pediatr Hematol Oncol 2000 22:550 [Google Scholar]
[18]. Weatherall DJ, Clegg JB, Inherited haemoglobin disorders: an increasing global health problemBull World Health Organ 2001 79:704-12. [Google Scholar]
[19]. WHO. Guidelines for the control of haemoglobin disorders. Report of the VIth Annual Meeting of the WHO Working 528 INDIAN J MED RES, 2011 Group on Haemoglobinopathies. Cagliairi, Sardinia, Geneva: World Health Organization; 1989 [Google Scholar]
[20]. Olivieri NF, Pakbaz Z, Vichinsky E, Hb E/beta-thalassaemia: a common & clinically diverse disorderIndian J Med Res 2011 134:522-31. [Google Scholar]
[21]. Weatherall DJ, Clegg JB, The Thalassaemia syndromes 2001 4th edOxford, U.K.Blackwell Science Ltd [Google Scholar]
[22]. Pathak MS, Borah MS, Kalita D, Disorders of haemoglobin variants in paediatric patients attending in a tertiary care hospital of north east IndiaInt J Biol Med Res 2014 5:3841-46. [Google Scholar]
[23]. Baruah MK, Saikia M, Baruah A, Pattern of haemoglobinopathies and thalassaemias in upper Assam region of North Eastern India: high performance liquid chromatography studies in 9000 patientsIndian J Pathol Microbiol 2014 57:236-43. [Google Scholar]
[24]. Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, Mahanta J, Prevalence of β-thalassaemia and other haemoglobinopathies in six cities in India: a multicentre studyJ Community Genet 2013 4:33-42. [Google Scholar]
[25]. Weatherall DJ, The challenge of haemoglobinopathies in resource poor countriesBr J Haematol 2011 154:736-44. [Google Scholar]
[26]. Chandrashekar V, Soni M, Haemoglobin Disorders in South IndiaISRN Hematol 2011 2011:748939 [Google Scholar]