Idiopathic Pulmonary Fibrosis and Myasthenia Gravis: An Unusual Association
Bharti Chogtu1, Daliparty Vasudev Malik2, Rahul Magazine3
1 Associate Professor, Department of Pharmacology, Kasturba Medical College, Manipal University, Manipal, Karnataka, India.
2 Postgraduate Student, Department of Pulmonary Medicine, Kasturba Medical College, Manipal University, Manipal, Karnataka, India.
3 Professor, Department of Pulmonary Medicine, Kasturba Medical College, Manipal University, Manipal, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Rahul Magazine, Professor, Pulmonary Medicine, Kasturba Medical College, Manipal University, Manipal-576104, Karnataka, India.
E-mail: rahulmagazine@gmail.com
Idiopathic Pulmonary Fibrosis (IPF) is a chronic fibrosing lung condition with high morbidity and mortality, accounting for about 25% of the cases of interstitial lung diseases. It usually has a progressive course resulting in death due to respiratory failure. Myasthenia Gravis (MG) is an autoimmune neuromuscular disease, caused by antibody mediated activity against acetylcholine receptor at the neuromuscular junction. It is characterized by fluctuating muscle weakness and fatigue. Extensive literature search did not reveal any case report of an association between these two conditions. Here we present a case of a patient with IPF who also developed MG. The diagnosis of IPF was based on High Resolution Computed Tomography (HRCT) of the lung and that of MG was based on clinical criteria and electrophysiological testing. The case was successfully managed.
Interstitial pneumonia, N-acetyl cysteine, Repetitive nerve stimulation
Case Report
A 54-year-old housewife presented to the chest outpatient department with a history of dry cough and exertional breathlessness from six years and recent onset of bilateral upper and lower limb weakness. She was diagnosed as Idiopathic Pulmonary Fibrosis (IPF) on the basis of High Resolution Computed Tomography (HRCT) of thorax for which she was initially started on a combination of steroids, N-acetyl cysteine (NAC) and azathioprine (triple therapy). But, as she developed cutaneous hypersensitivity to azathioprine, the drug was stopped and the patient was continued on NAC and steroids. The patient was also taking oral hypoglycaemic agents for steroid induced diabetes mellitus. She was a non-smoker and did not consume alcohol. On general physical examination, pulse rate was 70/min, regular, good volume and all the peripheral pulses were palpable. Respiratory rate was 22/min and blood pressure 140/90 mmHg, and she was afebrile. SpO2 at rest was 96-98% on room air. As none of the subsequent blood pressure recordings were above 126/80 mm Hg, the patient was not considered a hypertensive and not started on pharmacotherapy. The patient was able to cover a 6 minute walk distance of 378 meters without any symptoms or desaturation. Examination of the respiratory system revealed bilateral diffuse fine inspiratory crepitations on auscultation. Neurological examination revealed normal cranial nerve examination. Higher mental functions, sensory examination, deep tendon reflexes and cerebellar functions were normal with grade 5 power in all limbs. Limb weakness worsened following exercise testing. Examination of other systems did not reveal any abnormality.
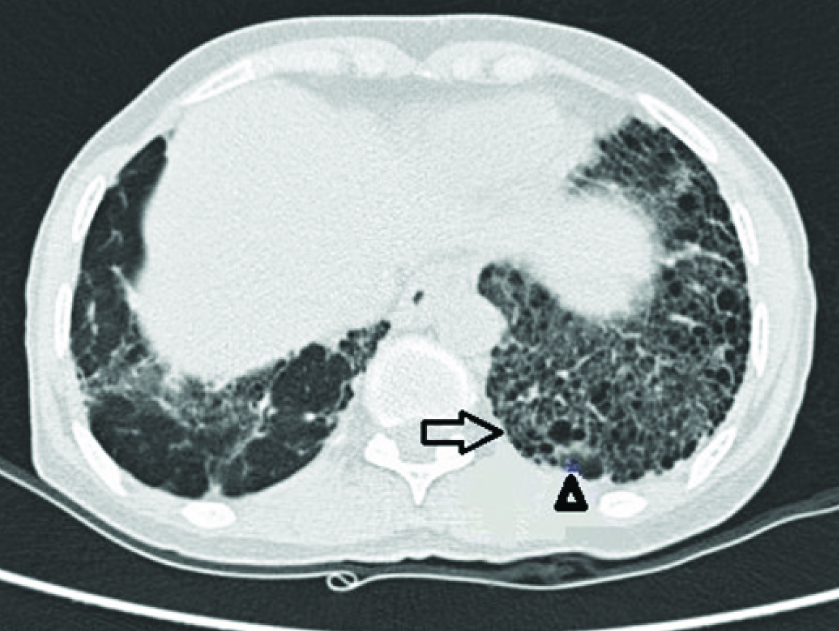
Haemogram showed normocytic normochromic anaemia (Hb=11.8 gm/dL), total leukocyte count of 12,100/mm3 with 71% neutrophils, 20% lymphocytes, and 1% eosinophils. Erythrocyte sedimentation rate was 61 mm in the first hour. Serum potassium level was 4.4 mmol/lit. Creatine kinase levels were low and showed a downward trend on serial monitoring. Anti-nuclear antibody and rheumatoid factor was negative. Serological testing for human immunodeficiency virus, sputum smear for acid fast bacilli was negative. Electrocardiogram, thyroid functions and liver functions were normal. Spirometry showed a restrictive pattern. Chest roentgenography revealed bilateral reticular opacities with honeycombing appearance, predominantly in the middle and lower zones. HRCT of the thorax was suggestive of Usual Interstitial Pneumonia (UIP) pattern [Table/Fig-1] and did not show any abnormality of the thymus gland. Magnetic Resonance Imaging (MRI) of the spine did not show any evidence of cord compression. Patient was followed up to check for progression of neurological symptoms. After one month, patient complained aggravation of weakness and nerve conduction study showed typical decrement of response in the right median nerve, left median nerve, facial nerve and accessory nerve to trapezius. Repetitive Nerve Stimulation (RNS) test was performed which yielded a decrement in compound muscle action potential amplitude of more than 10 percent, thus indicating a positive RNS test. Acetylcholine receptor (AchR) auto antibodies levels were normal. Based on the clinical features and a positive RNS test, the clinical diagnosis of myasthenia gravis was considered.
HRCT showing interstitial thickening (arrow) and honey combing (arrowhead) in the basal regions of the lung.
Patient was started on pyridostigmine for MG at a total daily dose of 120 mg over 4 divided doses. For IPF, patient was continued on a dose of methyl prednisolone 16 mg once daily (after azathioprine was withdrawn). Steroid dosage was tapered and adjusted based on the clinical condition of the patient. Patient is currently on prednisolone at a dose of 5 mg daily and on a dose of pyridostigmine 30 mg thrice daily. Patient has been on regular follow up once every 2 weeks and is currently stable. The timely treatment of myasthenia gravis has ensured that it does not have any deleterious effect on her respiratory condition. Informed consent to report the case was taken by the authors.
Discussion
Idiopathic pulmonary fibrosis is a disease affecting lungs that is characterized by progressive lung fibrosis. It has an estimated incidence of 7 to 17 per 100000 person years with a prevalence of 20-60 per 100000 person years as per the western scenario [1]. Inspite of the recent rise in IPF in India, the incidence and prevalence of the disease in India is difficult to determine as there are very limited epidemiological studies available [2]. For definitive diagnosis of IPF, a surgical lung biopsy is needed, but in this patient, a diagnosis was made in view of the clinical features and typical HRCT findings of areas of interstitial thickening and honey combing in basal regions [3]. On the other hand, myasthenia gravis is a progressive autoimmune disorder in which antibodies are directed against the neuromuscular junction. The annual incidence of MG is 3-4 cases per million and has a prevalence of about 60 cases per million in the general population [4]. There are no widely accepted diagnostic criteria or guidelines for diagnosis of myasthenia gravis. Clinical features, serological testing and electrophysiological studies are used to support the diagnosis. About 10-20% of generalized myasthenia gravis can have negative AChR antibody, as was the case in our patient. In such cases, anti muscle-specific kinase serology testing is to be done. However this facility is not available in our set up. However, if serology is inconclusive, then electrophysiological testing i.e., RNS is done. RNS test has a specificity of 97% for myasthenia gravis [5]. Though it can sometimes be negative, RNS testing has a crucial role in diagnosing seronegative myasthenia gravis [6]. A positive RNS test and improvement of muscular weakness with pyridostigmine lead to the diagnosis of myasthenia gravis in this patient even though AChR antibodies were negative. A variety of neuromuscular conditions can cause bilateral upper and lower limb muscle weakness. In this case, a diagnosis of Lambert Eaton myasthenic syndrome was excluded as improvement of muscular condition was not seen with exercise. A diagnosis of steroid induced myopathy was ruled out as the patient’s condition worsened after stopping steroids. Since the patient had none of the characteristic skin lesions and in view of the normal values of creatine kinase enzyme on serial monitoring, polymyositis and dermatomyositis were ruled out. HIV and hypothyroidism as causes of muscular weakness were ruled out in view of negative serology. Inspite of normal AChR autoantibody levels, the clinical features and a positive RNS test pointed to myasthenia gravis and the improvement of the patient’s condition with pyridostigmine pointed to the current diagnosis.
Co-existing IPF and myasthenia gravis can have a detrimental effect on the health of the patient as the respiratory manifestations of IPF can be compounded by the paralytic element of myasthenia gravis. However effective treatment of myasthenia gravis can avoid such a complication.
Concomitant IPF and myasthenia gravis increases patients’ morbidity. The respiratory condition of a patient of IPF can be complicated due to the added paralytic component of myasthenia gravis. As steroid dosage was tapered in this patient in view of the clinical improvement in terms of IPF, we can assume that this lead to the development of MG symptoms.
Azathioprine might be of benefit in cases of associated ILD and MG, but as the patient developed cutaneous hypersensitivity, its use was discontinued. Such an association may lead to multiple drugs being used for the management of different conditions, such as use of antibiotics (such as aminoglycosides) for treatment of chest infection in IPF. Care should be taken to avoid drugs such as aminoglycosides, anticonvulsants such as phenytoin and barbiturates as they affect neuromuscular junction transmission in certain cases. Since there were no clear cut reports of any association between IPF&MG, as IPF does not involve autoimmune processes in its etiology like MG, this association might just be a coincidence in our view.
Conclusion
Association of IPF and myasthenia gravis is rare and management of each condition requires careful drug selection to avoid adverse effects on the associated condition.
[1]. Ryu JH, Moua T, Daniels CE, Hartman TE, Yi ES, Utz JP, Idiopathic Pulmonary Fibrosis: Evolving ConceptsMayo Clin Proc 2014 89:1130-42. [Google Scholar]
[2]. Richeldi L, Rubin AS, Ardeev S, Udwadia ZF, Xu ZJ, Idiopathic Pulmonary Fibrosis in BRIC countries: the cases of Brazil, Russia, India, ChinaBio Med Central 2015 13:237 [Google Scholar]
[3]. Meltzer EB, Noble PW, Idiopathic pulmonary fibrosisOrphanet J Rare Dis 2008 3:8 [Google Scholar]
[4]. Mehndiratta MM, Pandey S, Kuntzer T, Acetylcholinesteraseinhibitor for myasthenia gravisCochrane Database Syst Rev 2014 Art.No.: CD006986. DOI: 10.1002/14651858.CD006986.pub3 [Google Scholar]
[5]. Corse A, Mankodi A. Myasthenia gravis. ePocrates online. Diseases page. Myasthenia gravis. Available at http://www.epocrates.com/. Last updated 1/2/2014. Accessed 1/16/2016 [Google Scholar]
[6]. Caliandro P, Evoli A, Stalberg E, Granata G, Tonali P, Padua L, The difficulty in confirming myasthenia gravis in a seronegativepatient: a possible neurophysiological approachNeuromuscul Disorder 2009 19(12):825-27. [Google Scholar]