Familial Constitutional Rearrangement of Chromosomes 4 & 8: Phenotypically Normal Mother and Abnormal Progeny
Fulesh Kunwar1, Sonal R. Bakshi2
1 Ph.D. Pursuing, Institute of Science, Nirma University, Sarkhej-Gandhinagar Highway, Ahmedabad, India.
2 Assistant Professor, Institute of Science, Nirma University, Sarkhej-Gandhinagar Highway, Ahmedabad, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sonal R. Bakshi, Assistant Professor, Institute of Science, Nirma University, Sarkhej-Gandhinagar Highway, Ahmedabad - 382 481 Gujarat, India.
E-mail: sonal.bakshi@nirmauni.ac.in
Balanced chromosome translocations carriers mostly do not have recognizable phenotypic expression but may have more risk of recurrent spontaneous abortions &/or children with serious birth defects due to unbalanced chromosome complements. Unbalanced chromosomal rearrangements have variable clinical expression and are rare. We present here a case report of three siblings affected with intellectual disability and minor dysmorphic features of face and limbs, born to a non-consanguineous couple in which mother had 5 abortions. The constitutional chromosome analysis revealed balanced translocation t (4;8) in mother and all the three siblings were karyotypically normal. Chromosomal microarray in one of the probands revealed partial monosomy 8pter-p23 and a partial trisomy 4pter-p16. Phenotypic features were recorded in 3 probands using Human Phenotype Ontology terms to query web-based tool Phenomizer. The harmonized description using globally accepted ontology is very important especially in case of rare genetic conditions and the heterogeneous phenotypes which make it even more challenging. The prevalence of sub-microscopic unbalanced translocations may be under-reported due to lesser use of molecular genetic analysis. The familial expression of abnormal phenotypes including intellectual disability make the individuals candidate for molecular genetic analysis and phenotyping to help defer the status of idiopathic mental retardation and identify sub-entity of genetic condition.
Chromosomal microarray, Familial constitutional t(4;8), Intellectual disability, Multiple abortions, Phenomizer
Case Report
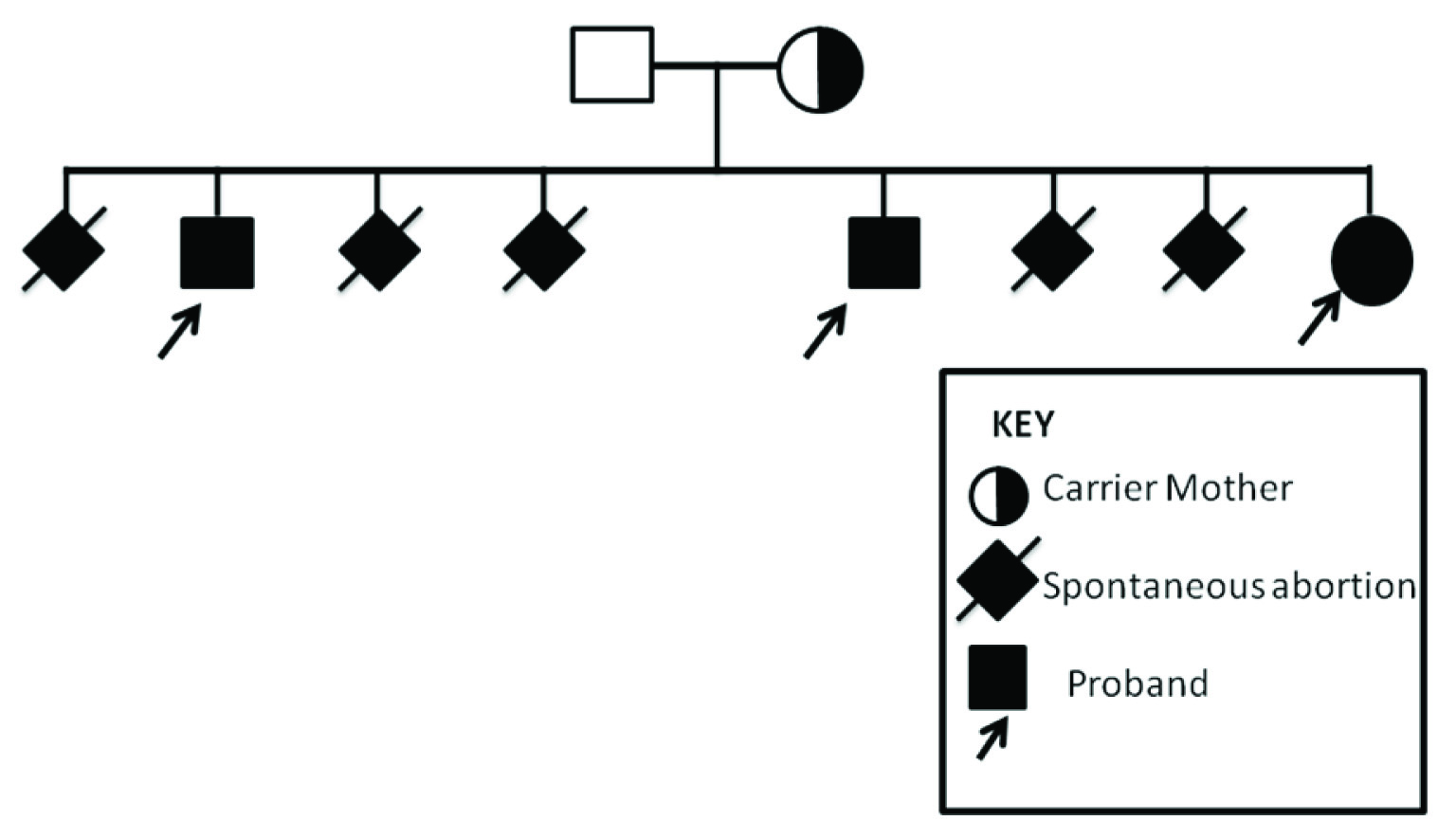
We describe here a familial case of idiopathic intellectual disability with low IQ (29% - 35%) based on BKT (Binet Kamat Intelligence Test) and minor dysmorphic features in three siblings born in a non-consanguineous couple with a history of 5 miscarriages [Table/Fig-1]. There was no remarkable history of exposure or illness during pregnancy and post birth. The phenotypic features of three siblings (23/M, 19/M and 9/F) are shown in [Table/Fig-2].
Pedigree of the siblings.
The Deep phenotypic features of siblings.

The case research was prospectively reviewed and approved by a duly constituted Institutional Human ethical committee.
Karyotyping: Standard GTG banding karyotyping procedure was done in siblings and parents after obtaining written informed consent. Karyotype was done as per the ISCN (International System for Human Cytogenetic Nomenclature-2013) [1] using digital image analysis system IKAROS (Metasystems, Germany).
CMAHD (Chromosomal Microarray-High Density): For further characterisation CMAHD (Whole genome oligonucleotide array analysis) was done in one of the three siblings (Proband-2) using lymphocyte genomic DNA. The assay includes high resolution molecular cytogenetic analysis with its 2,690,000 marker content {750,000 polymorphic (SNP) and 1,900,000 non-polymorphic (CNV) markers}. The microchip array used was CytoScan-HD (Affymetrix) for whole genome scan. The array can detect gains and losses at a minimum of 400kb and 200kb, respectively, across the genome, or smaller (≥50Kb) for clinically relevant deletion/duplication syndromes in the subtelomere and pericentromere region or targeted genes.
Phenomizer query: Phenotypic features in terms of minor/major abnormalities were recorded and entered in web-based tool Phenomizer [2] [Table/Fig-3]. Variations in body parts with respect to standard were also measured as shown in [Table/Fig-4].
Various phenotypic traits with variation or minor dysmorphic features of siblings using standard terminology.
| Proband-1 (09/F) | Proband-2 (19/M) | Proband-3 (23/M) |
|---|
| Hair pattern | Normal | Normal | Single whorl at abnormal position |
| Skull | High anterior hairline, Broad and large forehead | Microcephaly | Normal |
| Shape of face | Square face, Pointed chin | Abnormality of the zygomatic arch | Square face, Chin dimple |
| Tongue | Microglossia | Bifid tongue, Broad uvula | Normal |
| Nose | Wide nose, Long nose, Wide nasal bridge | Asymmetry of the nares, Low hanging columella, Abnormality of the nasal dorsum | Short nose, Bulbous nose Hypoplastic nasal alae, Broad nasal tip, Flared nostrils |
| Lips | Thin vermilion border of upper lip, Absent cupid’s bow, Everted lower lip vermilion | Curved linear dimple below the lower lip | Normal |
| Ears | Prominent antitragus, Broad width of superior crus of antihelix, Macrotia | Low-set ears, Abnormality of the tragus, Everted antitragus, Underdeveloped antitragus, Attached earlobe, Angulated antihelix, Abnormality of superior crus of antihelix, Macrotia | Macrotia |
| Eyes | Strabismus | Strabismus, Up-slanted palpebral fissure | Up-slanted palpebral fissure, Strabismus |
| Periorbital Region | Telecanthus | Telecanthus | Telecanthus, Epicanthus |
| Philtrum | Broad philtrum | Smooth philtrum, Malaligned philtral ridges | Broad philtrum |
| Palate | Normal | High palate | Normal |
| Neck | Short | Normal | Normal |
| Palm | Broad palm | Broad palm, Abnormality of the thenar eminence | Normal |
| Fingers | Clinodactyly of the 5th finger | Normal | Camptodactyly of finger |
| Nails | Leukonychia | Normal | Normal |
| Legs | Normal | Short lower limbs | Normal |
| Feet | Normal | Broad foot | Normal |
| Toes | Normal | Widely spaced toes | Normal |
| Chest | Barrel-shaped chest | Barrel-shaped chest | Normal |
Variations in body parts with respect to standard measurement (N=Normal, V=Variation).
| Body Parts | Proband-(109/F) | Proband-(219/M) | Proband-(323/M) |
|---|
| Head: SkullHair | VN | VN | VV |
| Nose | V | V | V |
| Neck | V | N | N |
| Chest appearance | V | V | V |
| Nails: FingerToes | VN | NV | VN |
| Head CircumferenceChest CircumferenceShoulder Width | NVV | VVV | NVV |
| Upper LimbsFull Hand LengthPalm LengthMiddle Finger Length | NNNN | NVVV | NVVV |
| Foot Length | N | N | V |
| Outer CanthalInner CanthalInter PapillaryPalpebral FissurePhiltrumEar Length | VVVV–V | VVVVVV | VVVVVV |
Discussion
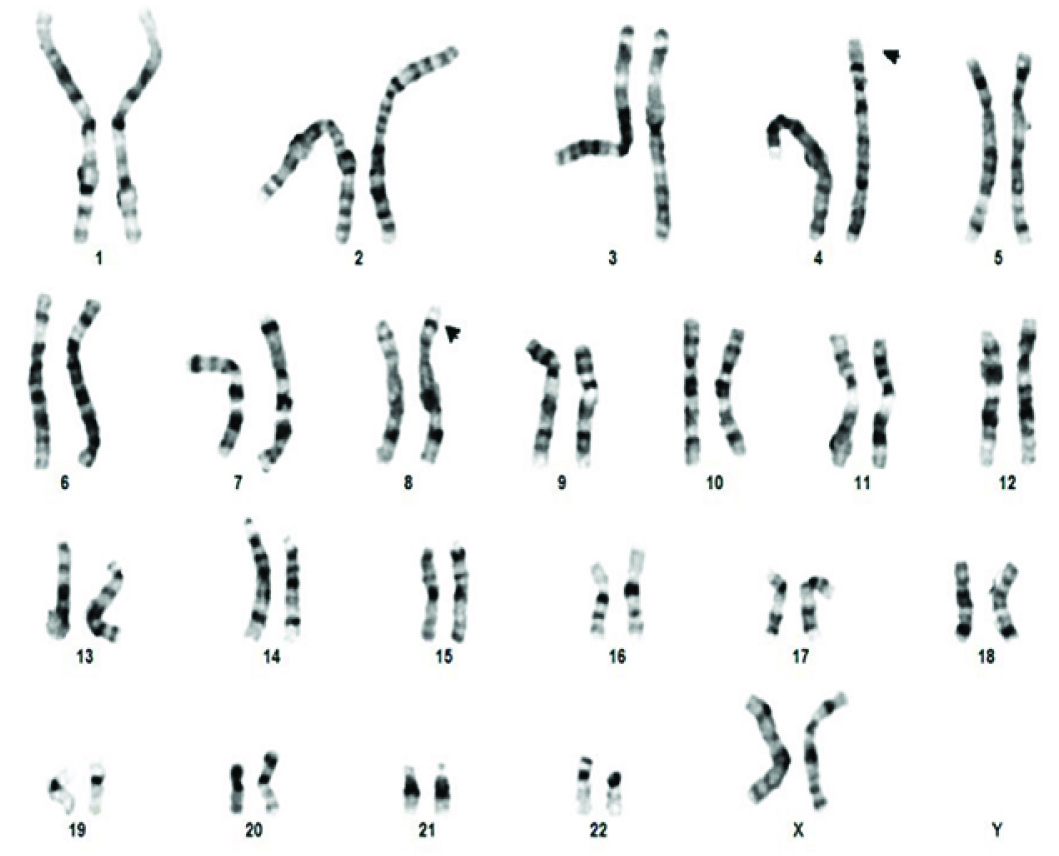
The high resolution karyotypes of all the three siblings and father were normal at 550 band level whereas the mother had balanced t(4p;8p) as shown in [Table/Fig-5]. The CMHD result of PB-2 was arr4p16.3p16.1 (68,345-9,520,844)x3, 8p23.3p23.1(158,048-6,982,980)x1. There was an approximately 9.4 Mb gain of the terminal short arm of chromosome 4 and a 6.8 Mb loss of the terminal short arm of chromosome 8. The pattern of imbalance observed in this patient was consistent with an unbalanced chromosome complement resulting from a maternal balanced translocation between the short arms of chromosome 4 and chromosome 8.
Karyotype of mother showing t(4p;8p).
Phenomizer scores indicated the most likely diagnoses as Simosa Craniofacial Syndrome (2.5838, 2.8056, 2.1902) that was common for all the three siblings. The results in one female (Proband-1) and one male patient (Proband-2) included four common genetic conditions [Table/Fig-6], in addition to others. The Phenomizer query result did not indicate t(4p;8p) or loss of 8p which may be due to non classical or variant phenotypic features.
Result generated by web based software Phenomizer for all the three siblings respectively showing ranked list of possibilities and associated gene based on score
| Score | Disease entry | KnownGenes | Score | Disease entry | KnownGenes | Score | Disease entry | KnownGenes |
|---|
| 2.5838 | 182150 SIMOSACRANIOFACIALSYNDROME(OMIM:182150) | | 3.0583 | #212720 MARTSOLFSYNDROME;;CATARACTMENTALRETARDATION-HYPOGONADISM(OMIM:212720) | RAB3GAP2 | 2.4526 | %277720 WHISTLINGFACE SYNDROME, RECESSIVEFORM (OMIM:277720) | |
| 2.5751 | #300321 FG SYNDROME2; FGS2 (OMIM:300321) | FLNA | 2.8094 | FACIOGENITALDYSPLASIA(OMIM:305400) | FGD1 | 2.3832 | #614756 CEREBELLARATAXIA, NONPROGRESSIVE,WITH MENTAL RETARDATION;CANPMR (OMIM:614756) | CAMTA1 |
| 2.5334 | %602342 PLANTARLIPOMATOSIS,UNUSUAL FACIES, ANDDEVELOPMENTAL DELAY;;PIERPONT SYNDROME(OMIM:602342) | | 2.8056 | 182150 SIMOSACRANIOFACIALSYNDROME(OMIM:182150) | | 2.3712 | #193700 ARTHROGRYPOSIS,DISTAL, TYPE 2A; DA2A;;FREEMAN-SHELDONSYNDROME;FSS;;WHISTLING FACE-WINDMILLVANE HAND SYNDROME;;CRANIOCARPOTARSALDYSTROPHY;;CRANIOCARPOTARSALDYSPLASIA(OMIM:193700) | MYH3 |
| 2.4915 | %164220 SCHILBACH-ROTT SYNDROME;;OCULAR HYPOTELORISM,SUBMUCOSAL CLEFTPALATE, AND HYPOSPADIAS;;CLEFT PALATE,HYPOTELORISM, ANDHYPOSPADIAS;;BLEPHAROFACIOSKELETALSYNDROME; BRSS(OMIM:164220) | | 2.7673 | 606155 FRYNS-AFTIMOSSYNDROME;;PACHYGYRIA,MENTAL RETARDATION,EPILEPSY, ANDCHARACTERISTICFACIES;; CEREBROOCULOFACIALLYMPHATIC SYNDROME;;COFL SYNDROME;;MENTALRETARDATIONWITH EPILEPSYAND CHARACTERISTICFACIES (OMIM:606155) | | 2.2532 | #300749 MENTALRETARDATION ANDMICROCEPHALYWITH PONTINE ANDCEREBELLARHYPOPLASIA;MICPCH;;MICPCHSYNDROME;;MENTALRETARDATION, XLINKED,SYNDROMIC, NAJMTYPE; MRXSNA(OMIM:300749) | CASK |
| 2.4834 | FACIOGENITAL DYSPLASIA(OMIM:305400) | FGD1 | 2.7441 | #158170 CHROMOSOME9P DELETION SYNDROME;;MONOSOMY 9P SYNDROME(OMIM:158170) | | 2.1991 | 612948 STARGARDTMACULAR DEGENERATION,ABSENT OR HYPOPLASTIC CORPUSCALLOSUM,MENTALRETARDATION, ANDDYSMORPHIC FEATURES(OMIM:612948) | |
| 2.3693 | %227330 FACIODIGITOGENITALSYNDROME, AUTOSOMALRECESSIVE;;AARSKOG-LIKESYNDROME;;KUWAIT TYPEFACIODIGITOGENITALSYNDROME (OMIM:227330) | | 2.6672 | %227330 FACIODIGITOGENITALSYNDROME, AUTOSOMALRECESSIVE;;AARSKOG-LIKESYNDROME;;KUWAIT TYPEFACIODIGITOGENITALSYNDROME (OMIM:227330) | | 2.1902 | 182150 SIMOSACRANIOFACIALSYNDROME(OMIM:182150) | |
| 2.3663 | KBG SYNDROME(OMIM:148050) | ANKRD11 | 2.6447 | %602342 PLANTARLIPOMATOSIS, UNUSUALFACIES, AND DEVELOPMENTALDELAY;; PIERPONT SYNDROME(OMIM:602342) | | 2.1806 | #613544 CHROMOSOME6Q11-Q14 DELETIONSYNDROMECHROMOSOME6Q13-Q14 DELETIONSYNDROME, INCLUDED(OMIM:613544) | |
| 2.3270 | 311450 PALLISTER WSYNDROME;;WSYNDROME(OMIM:311450) | | 2.6052 | #610536 MANDIBULOFACIALDYSOSTOSIS,GUION-ALMEIDA TYPE;MFDGA;;MANDIBULOFACIALDYSOSTOSISWITH MICROCEPHALY;MFDM;;GROWTHAND MENTAL RETARDATION,MANDIBULOFACIALDYSOSTOSIS, MICROCEPHALY,AND CLEFT PALATE(OMIM:610536) | EFTUD2 | 2.1623 | #300143 MENTALRETARDATION, X-LINKED21; MRX21;;MENTALRETARDATION, XLINKED34; MRX34 (OMIM:300143) | IL1RAPL1 |
| 2.2904 | 606155 FRYNS-AFTIMOSSYNDROME;; PACHYGYRIA,MENTAL RETARDATION,EPILEPSY, ANDCHARACTERISTIC FACIES;;CEREBROOCULOFACIALLYMPHATIC SYNDROME;;COFL SYNDROME;;MENTAL RETARDATIONWITH EPILEPSYAND CHARACTERISTICFACIES (OMIM:606155) | | 2.5781 | %181270 SCALP-EAR-NIPPLESYNDROME;;FINLAY-MARKSSYNDROME;; SENSYNDROME (OMIM:181270) | KCTD1 | 2.1617 | RING CHROMOSOME 1(ORPHANET:1437) | |
| 2.2733 | %145420HYPERTELORISM,TEEBITYPE;;BRACHYCEPHALOFRONTONASALDYSPLASIA(OMIM:145420) | | 2.5034 | #610954 PITT-HOPKINSSYNDROME; PTHS;;ENCEPHALOPATHY,SEVERE EPILEPTIC,WITH AUTONOMICDYSFUNCTION;;MENTAL RETARDATION,SYNDROMAL,WITH INTERMITTENTHYPERVENTILATION(OMIM:610954) | TCF4 | 2.1582 | #611091 MENTAL RETARDATION,AUTOSOMALRECESSIVE 5; MRT5 (OMIM:611091) | NSUN2 |
Phenotypic expression in segmental aneuploidy generally varies depending on the size of the chromosomal region involved. Genetic factor such as abnormal constitutional karyotype of parental origin or de novo are the reasons for spontaneous abortion. Carriers of balanced chromosomal structural rearrangements are without remarkable phenotypic signature and thus inherited familial cases through the multiple generations can go without detection. The chromosomal translocation involving 4p and 8p has been reported earlier which are mainly associated with partial trisomy 8p and partial monosmy 4p [3,4], however the reverse is of rare occurrence. Subjects with deletion in 4p; der(4) are reported to have Wolf-Hirschhorn syndrome, whereas cases with deletion at 8p; der(8) are reported to have a milder spectrum of dysmorphic features [3]. There is a wide spectrum of clinical features in people having unbalanced t(4;8) which makes identification of these patients very challenging [5].
Various cryptic translocations can be missed by cytogenetic banding techniques mainly because banding pattern and size are almost similar for both the regions at the 400-500 band resolution. Hence, similar to the widely reported cryptic t(11q;22q); the t(4p;8p) may also be a recurrent translocation [6].
The breakpoint reported in 8p region is within olfactory receptor gene cluster whereas there are two different breakpoints generally reported in 4p; at a distance of approximately 5 and 14Mb from the telomere which may account for the spectrum of phenotypic features that vary with the size of chromosomal region involved [7]. The reports of deletion of 8p generally include common clinical features like developmental delay, low birth weight, congenital heart disease, and a characteristic behaviour like hyperactivity and impulsiveness. The cardiac anomaly reported in these cases is atrioventricular septal defect which could be due to deletion of GATA4 gene present on 8p [8]. But not all the patients have congenital heart defects, the possible reason may be compensatory increases in GATA5 or GATA6 which may mitigate the effects of GATA4 deletion and haploin sufficiency for other cardiac transcription factor genes (e.g. TBX5, NKX2–5) which causes congenital heart disease [8]. The patients having intellectual disability generally also show growth retardation but people with a gain at 4p due to the unbalanced t(4p;8p) are generally tall. This could probably be due to the presence of third copy of FGFR3 gene on the 4p16.3 [9].
According to the Unique database only few cases of der(8) have been reported in medical literature (Unique) [10].
The unbalanced translocation resulting in both partial trisomy for 4p16-pter and partial monosomy for 8p23-pter can be associated with the maternal constitutional t(4;8) (p16;p23). A chromosomal break can cause loss of function of gene if it disrupts the coding sequence or if it separates the coding sequence with cis-acting regulatory sequence. A breakpoint can provide a valuable clue to exact physical location of disease gene. In our case the genetic aberration in Proband-2 involving loss in 8p, the breakpoint region is at 6,982,980 position which occurs between two genes DEFA1 and DEFT1P, probably in the non-coding region but might have affected function of these genes. The gain in 4p might have lead to higher copy number of genes including OR7E83P and LOC101928948 along with others as the breaking region is at 9,520,844 position which occur in between the above two genes [11]. This genomic imbalance observed using CMA may be responsible for congenital minor dysmorphisms and intellectual disabilities due to the dosage effect of genes involved. The rest of the two probands with similar clinical presentation are also likely to carry the similar genomic aberrations though normal at GTG banded karyotype level. The recurrent 8p rearrangements can also occur as a consequence of an inversion polymorphism mediated by two olfactory receptor (OR) gene clusters which can be of parental origin. The olfactory receptor gene clusters exist at 4p16, as well as 8p23 which may be implicated in the genesis of the recurrent t(4;8)(p16;p23) [3]. Giglio S et al., hypothesized that the heterozygous submicroscopic inversions of both 4p and 8p could prevent correct synapsis of the inverted regions at meiosis. As a result, both tetrads might assume a configuration that could result in an illegitimate but “homologous” crossover between the OR-gene clusters present on both 4p and 8p i.e., the double heterozygous inversion will make two nonhomologous chromosomes available to recombine with each other [3].
The unbalanced t(4p;8p) is not always detectable by conventional cytogenetics/multi-colour Fluorescence in situ hybridization techniques [5,12], hence molecular cytogenetic techniques may be helpful to unravel unbalanced chromosomal translocations.
Conclusion
It is important to put on record every case of unbalanced t(4;8) along with detailed phenotypic data collected and described in a harmonized way. The high resolution genotyping data in the current era can be more meaningful if matched with phenotyping data collected in detail. The combined study of phenotypic and molecular genetics characterization in abnormal constitutional karyotype cases are required to be pooled to help delineate a subentity of genetic condition. Such efforts can help better clinical management and genetic counselling apart from the molecular pathogenesis studies.
[1]. Shaffer LG, McGowan-Jorda J, Schmid M, An International System for Human Cytogenetic Nomenclature 2013 BaselS. Karger AG:140 [Google Scholar]
[2]. Kohler S, Schulz MH, Krawitz P, Bauer S, Dolken S, Ott CE, Clinical diagnostics in human genetics with semantic similarity searches in ontologiesAm J Hum Genet 2009 85(4):457-64. [Google Scholar]
[3]. Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Heterozygous Submicroscopic Inversions Involving Olfactory Receptor–Gene Clusters Mediate the Recurrent t(4;8)(p16;p23) TranslocationAm J Hum Genet 2002 71(2):276-85. [Google Scholar]
[4]. Mau UA, Backsch C, Schaudt H, Trefz FK, Kaiser P, Three-year-old girl with partial trisomy 4p and partial monosomy 8p with resemblance to Brachmann-de Lange syndrome–another locus for Brachmann-de Lange syndrome on 4p?Am J Med Genet 2000 91(3):180-84. [Google Scholar]
[5]. Tonnies H, Stumm M, Neumann L, Volleth M, Grumpelt U, Musebeck J, Two further cases of WHS with unbalanced de novo translocation t(4;8) characterised by CGH and FISHJ Med Genet 2001 38(6):E21 [Google Scholar]
[6]. Kurahashi H, Shaikh TH, Emanuel BS, Alu-mediated PCR artifacts and the constitutional t(11;22) breakpointHum Mol Genet 2000 November 1 2000; 9(18):2727-32. [Google Scholar]
[7]. Zollino M, Lecce R, Selicorni A, Murdolo M, Mancuso I, Marangi G, A double cryptic chromosome imbalance is an important factor to explain phenotypic variability in Wolf-Hirschhorn syndromeEur J Hum Genet 2004 07/07/online 12(10):797-804. [Google Scholar]
[8]. Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, GATA4 haploin sufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart diseaseAm J Med Genet 1999 83(3):201-06. [Google Scholar]
[9]. Partington MW, Fagan K, Soubjaki V, Turner G, Translocations involving 4p16.3 in three families: deletion causing the Pitt-Rogers-Danks syndrome and duplication resulting in a new overgrowth syndromeJ Med Genet 1997 34(9):719-28. [Google Scholar]
[10]. http://www.rarechromo.org/information/Chromosome%20%208/4p%208p%20Translocation%20FTNW.pdf accessed on 05.09.2014 [Google Scholar]
[11]. http://www.ncbi.nlm.nih.gov accessed on 29.09.2015 [Google Scholar]
[12]. Skrlec I, Wagner J, Pubeljic S, Heffer M, Stipoljev F, De novo case of a partial trisomy 4p and a partial monosomy 8pColl Antropol 2014 38(1):319-23. [Google Scholar]