Fine Needle Aspiration (FNA) is a simple, inexpensive and innocuous diagnostic tool. It is increasingly accepted in daily medical practice. We report a case of Gaucher’s disease diagnosed by FNA of enlarged spleen in a seven-year-old male presented with complaints of fever, on and off irregular bowel habits, abdominal distension and asymmetric gaze for four years and decreased vision in night for last one year. The diagnosis was based on the finding of large, macrophage like cells with abundant, pale, fibrillary cytoplasm and small nuclei. The patient had no family history of Gaucher’s disease, and the diagnosis was not suspected clinically.
Case Report
A seven-year-old-male presented in the Paediatric-emergency–ward with complaints of fever, on and off irregular bowel habits, abdominal distension and asymmetric gaze for four years and decreased vision in night since last one year. Fever was of high grade, continuous in nature, not associated with chills, rigor or sweating and relieved by medication. He had irregular bowel habits, passed stool six to seven times per day. The stool was watery in consistency, mixed with mucus and froth with no blood. Abdominal distension was of insidious onset, progressive in nature and was associated with pain. There was no history of jaundice, seizure, bleeding, oliguria, haematuria, loss of appetite and swelling of hand, feet or face. Blood transfusion was never been done. Antenatal, natal and postnatal history was not relevant and he was immunized as per national immunisation schedule.
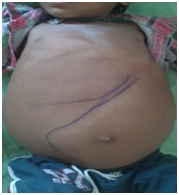
On clinical examination pallor, squint and bitot’s spot were found. Oral cavity examination revealed angular stomatitis and poor oral hygiene. Multiple, small, discrete and freely mobile, left posterior cervical, axillary and inguinal lymphnodes were found. Oedema, icterus, clubbing and cyanosis were absent. Spine was normal. Gait was wide based and lordotic. Abdominal examination revealed distention and umbilicus was shifted inferiorly. There was no scar mark, engorged vein, visible peristalsis or divarication of recti. Hernial orifices were intact. Scrotum was normal. Superficial palpation did not reveal any abnormality. Deep palpation revealed non-tender hepatomegaly and liver was palpable 5.5 cm below right subcostal margin in mid-clavicular line and spleen was 21 cm below left subcostal margin in right spinoumbilical line, crossing umbilicus [Table/Fig-1]. There was neither other palpable lump nor sign of hepatocellular failure. Ascites was absent. Peristaltic sound was 3-4/min. Arterial bruit and hepatic and splenic rub were absent. Examination of Central Vascular System, Respiratory System, and Central Nervous System did not reveal any abnormality except for the presence of bulging of lower sternum [Table/Fig-1] and squint. The differential diagnosis of kala-azar, malaria, and haemato-lymphoid malignancy were considered. The patient was investigated for Complete Blood Count, Peripheral Blood Smear Examination, Erythrocyte Sediment Rate, Liver Function Test, Kidney Function Test, iron studies, level of serum acid phosphatase and transcobalamine, Chest X-ray P-A view, X-ray of bilateral thigh and legs, Ultrasonogram of abdomen, Fundoscopy, CECT of brain and splenic aspiration.
Haematological examination revealed pancytopaenia and Erytherocyte Sedimentation Rate (ESR) of 90mm in the first hour. Peripheral Blood Smear was negative for malarial parasite and any abnormal cells. Biochemical examination revealed normal finding of liver and kidney function test but increased serum acid phosphatase, serum ferritin and transcobalamine II. Chest X Ray showed hilar lymphadenopathy. X-ray of long bones was found normal. USG abdomen revealed multiple enlarged, mesenteric lymphnodes, the largest measuring 1.2× 0.6 cm2 along with massive splenomegaly and moderate hepatomegaly. Fundoscopy showed optic pit with maculopathy. CECT brain showed normal scan.
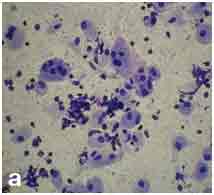
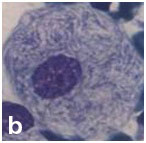
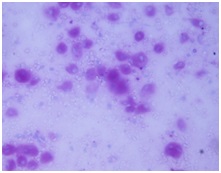
Cytological examination of splenic aspirate: Splenic aspiration was done with aseptic and antiseptic precaution. The wet smear was fixed in 95% ethanol and the dried smear was stained with May-Grunwald-Giemsa stain. The smear was cellular and showed large oval to polygonal macrophage like cells with central to eccentrically placed small nuclei and abundant amount of fibrillary cytoplasm having crumpled paper appearance along with its binucleated and multinucleated giant forms [Table/Fig-2a&b]. These cells were positive for Periodic-Acid–Schiff stain [Table/Fig-3]. Leishmania donovani (LD) bodies were not found.
Patient with hepatosplenomegaly.
Cellular smear showing macrophage like cells with binucleated and multinucleated giant forms (PAP stain; X100).
Macrophage like cell with small nuclei and abundant amount of fibrillary cytoplasm having crumpled paper appearance (PAP stain; X400).
Cells showing positivity for PAS stain.
On the basis of above findings, a diagnosis of non-neuronopathic type of Gaucher’s disease was made and clinical differential diagnosis of kala-azar, malaria and haematological malignancy were ruled out. The level of the lysosomal enzyme glucocerebrosidase was measured after the cytological diagnosis and was found to be reduced which confirmed the diagnosis.
The detailed family history was searched after the diagnosis was confirmed but did not reveal anything significant. The level of glucocerebrosidase among parents and siblings were found normal. Clinical confirmation of heterozygosity required the use of molecular biotechnology methods which was not available in our institute and hence not done. The case was referred to higher centre for management.
Discussion
Gaucher’s disease is the most common lysosomal storage disorder. It was identified by a French dermatologist, Phillipe Gaucher in 1882 [1]. It was found that a deficiency in the enzyme glucocerebrosidase (which breaks down the cell membrane component glucocerebroside), leads to an accumulation of glucocerebroside within the lysosomes of macrophages throughout the body [1,2]. It is a genetic disorder transmitted from parent to child [2]. Symptoms often appear at late age but no age is exempted [3]. An early onset of clinical sign and symptoms predispose patients to irreversible complications [3]. It has been found to be a progressive, debilitating, and sometimes life threatening disease [3].
It has been classified into: type 1(non-neuronopathic); type 2 (acute neuronopathic); and type 3 (subacute/chronic neuronopathic) [2–4]. Among these three, type 1 is the most common [2]. It has high prevalence among Ashkenazi Jews and rare in Indian subcontinent [5]. The patients remain asymptomatic or mildly symptomatic but severe disability with disfigurement and even death may occur [2]. Clinical features include unexplained spleno-hepatomegaly, anaemia, thrombocytopaenia and skeletal disease [1]. Most common debilitating feature is the insidious skeletal complications [2]. The present case was of seven-year-old and had the complaints of fever, on and off irregular bowel habits, abdominal distension and asymmetric gaze for 4 years and decreased vision in night for last 1 year. On clinical examination pallor, squint, bitot’s spot, angular stomatits, cervical and axillary lymphadenopathy, hepatosplenomegaly and bulging of lower sternum and squint were found.
The less common form of the diseases are type 2 and type 3, which are characterized by the presence of central nervous symptoms such as myoclonic seizures, oculomotoric apraxia and a slight mental retardation [2].
Apart from clinical features, morphologic, haematologic, and biochemical indicators can help to establish the diagnosis [2]. In the present case there was pancytopaenia, increased ESR, increased serum acid phosphatase, serum ferritin and transcobalamine II. The definitive diagnosis is made by determining the catalytic activity of the lysosomal enzyme glucocerebrosidase, in peripheral leukocytes in a suspected case of Gaucher’s disease [1,4]. Lack of clinical awareness and laboratory facility of the enzyme assay limit its application in clinical practice and many cases are diagnosed by bone marrow and liver biopsy [1]. In the present case clinical differential diagnosis of kala-azar, malaria and haematological malignancy was thought and level of the enzyme was measured after the cytological diagnosis and found to be reduced. About 150 mutations of the glucocerebrosidase gene have been identified in patients with Gaucher’s disease, some of which are predictive of phenotype [2].
Fine Needle Aspiration Cytology (FNAC) is a diagnostic tool and is increasingly accepted in daily medical practice [6]. It is useful for palpable and radiological accessible lesion and provides information about the cytological aspects of punctured site [6]. Cell block prepared from aspirated material has greatest diagnostic use and provides histological information [6]. The diagnostic cytological finding of Gaucher’s disease is the presence of large macrophage like cells with abundant, pale, fibrillary cytoplasm and small nuclei [7].
The present case had strong clinical suspicion of Kala-azar and splenic aspiration was advised because it is less painful and more sensitive procedure than bone marrow aspiration [8]. Sensitivity of splenic aspiration and bone marrow aspiration was found 93–99% and 53–86% respectively [9]. FNAC of lymph nodes were not attempted because of its smaller size and less sensitivity of the procedure (53–65%) [9]. The present case was diagnosed by finding classical Gaucher’s cell and the diagnosis of kala-azar was ruled out by absence of LD bodies.
Enzyme replacement, substrate reduction therapy, bone marrow transplantation, blood transfusion and surgery are the common modalities of the treatment [10].
Conclusion
Early diagnosis and treatment is the best way to prevent further progress of the disease. A multidisciplinary approach and regular follow-up will help a patient to lead a better life with less complications. Laboratory facilities for diagnosis and awareness in clinical practice is required to combat the condition.
[1]. Niederau C, Haussinger D, Gaucher’s disease: a review for the internist and hepatologistHepatogastroenterology 2000 47:984-97. [Google Scholar]
[2]. Fumić K, Stavljenić-Rukavina A, Mrsić M, Potocki K, Gaucher disease:diagnosis and treatmentActa Med Croatica 2004 58:353-58. [Google Scholar]
[3]. Gupta SS, Mondal P, Basu N, Mallick MG, Gaucher’s disease with uncommon presentationsJ Cytol 2009 26:117-19. [Google Scholar]
[4]. Niederau C, Rolfs A, Vom Dahl S, Häussinger D, Poll LW, Mengel E, Diagnosis and therapy of Gaucher disease. Current recommendations of German therapy centers in the year 2000Med Klin (Munich) 2001 96:32-39. [Google Scholar]
[5]. Bohra V, Nair V, Gaucher’s diseaseIndian J Endocrinol Metab 2011 15(3):182-86. [Google Scholar]
[6]. Ortiz J, Fernández D, Bullón A, Gaucher’s disease: morphological findings in a case studied with fine needle aspirationCytopathology 2002 13:371-74. [Google Scholar]
[7]. Domanski H, Dejmek A, Ljung R, Gaucher’s disease in an infant diagnosed by fine needle aspiration of the liver and spleen. A case reportActa Cytol 1992 36:410-12.18. [Google Scholar]
[8]. Sarkar CB, Alam KS, Jamal MF, Rahman S, Huq MH, Musa AK, Sensitivity of splenic and bone marrow aspirate study for diagnosis of kala-azarMymensingh Med J 2004 13:130-33. [Google Scholar]
[9]. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, Visceral leishmaniasis: what are the needs for diagnosis, treatment and control?Nature Reviews/Microbiology 2007 5:S7-16. [Google Scholar]
[10]. Rizk TM, Ariganjoye RO, Alsaeed GI, Gaucher disease. Unusual presentation and mini-reviewNeurosciences (Riyadh) 2015 20:271-76. [Google Scholar]