Pancreatoblastoma an Unusual Occurrence of a Tumour in Paediatric Age Group: A Case Report
Pankaj Halder1, Bedabrata Mukhopadhyay2, Chhanda Das3, Madhumita Mukhopadhyay4
1 Asssitant Professor, Department of Paediatric Surgery, Dr B C Roy Post Graduate Institute of Paediatic Sciences, Kolkata, West Bengal, India.
2 Post Graduate Student, Department of Biochemistry, IMS, BHU, Varanasi, Uttar Pradesh, India.
3 Assistant Professor, Department of Pathology, IPGME & R, India.
4 Professor, Department of Pathology, IPGME & R, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Chhanda Das, 31 Eastern Park, Ist Road, Santoshpur, Kolkata-70075, West Bengal, India.
E-mail: chhhdas@gmail.com
Pancreatoblastoma, is rare exocrine malignant tumour of childhood. We are reporting a case of three-year-old child presented to our hospital suffering from vague abdominal pain for further examination and treatment. Clinical examination revealed only a palpable abdominal mass. CT Scan revealed a huge complex space occupying lesion 9.1x8.8x9.2cm with large central cystic degeneration and lobulated enhancing peripheral solid components with foci of calcification, seem to arise from body and tail regions of pancreas. Surgery was done and mass was removed. By histopathology and immunohistochemistry it was diagnosed as pancreatoblastoma. The prognosis is very good in paediatric age, lacking evidence of metastatic disease at first presentation. Therefore early diagnosis is needed for specific treatment. The case is being reported because of its rarity.
Alphafetoprotein, Mesenteric cyst
Case Report
A three-year-old young female child presented in the department of paediatric surgery with chief complaints of gradual swelling in umbilical region and vague abdominal discomfort for last 3 months. She was single child, her medical history revealed nothing significant.
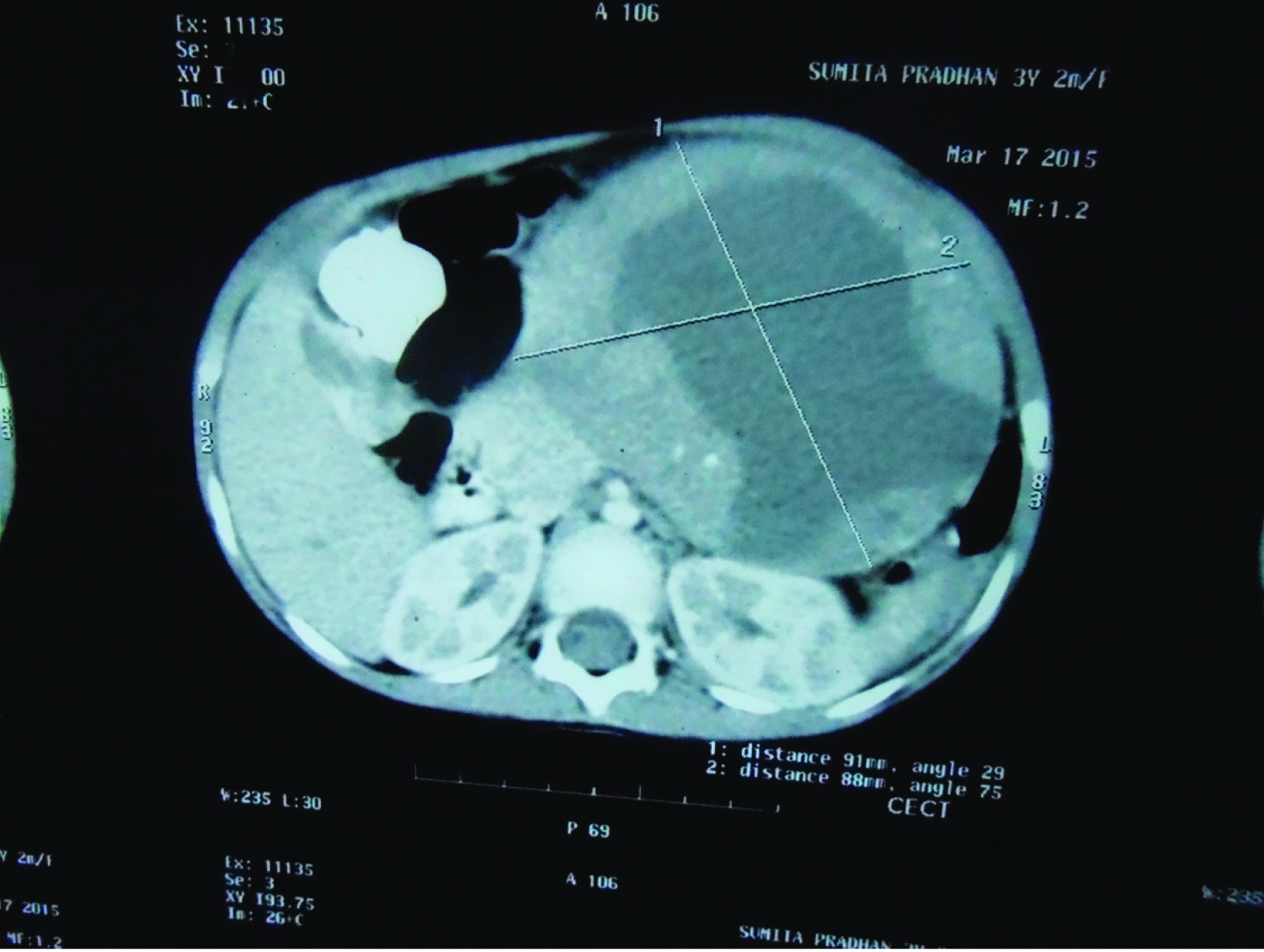
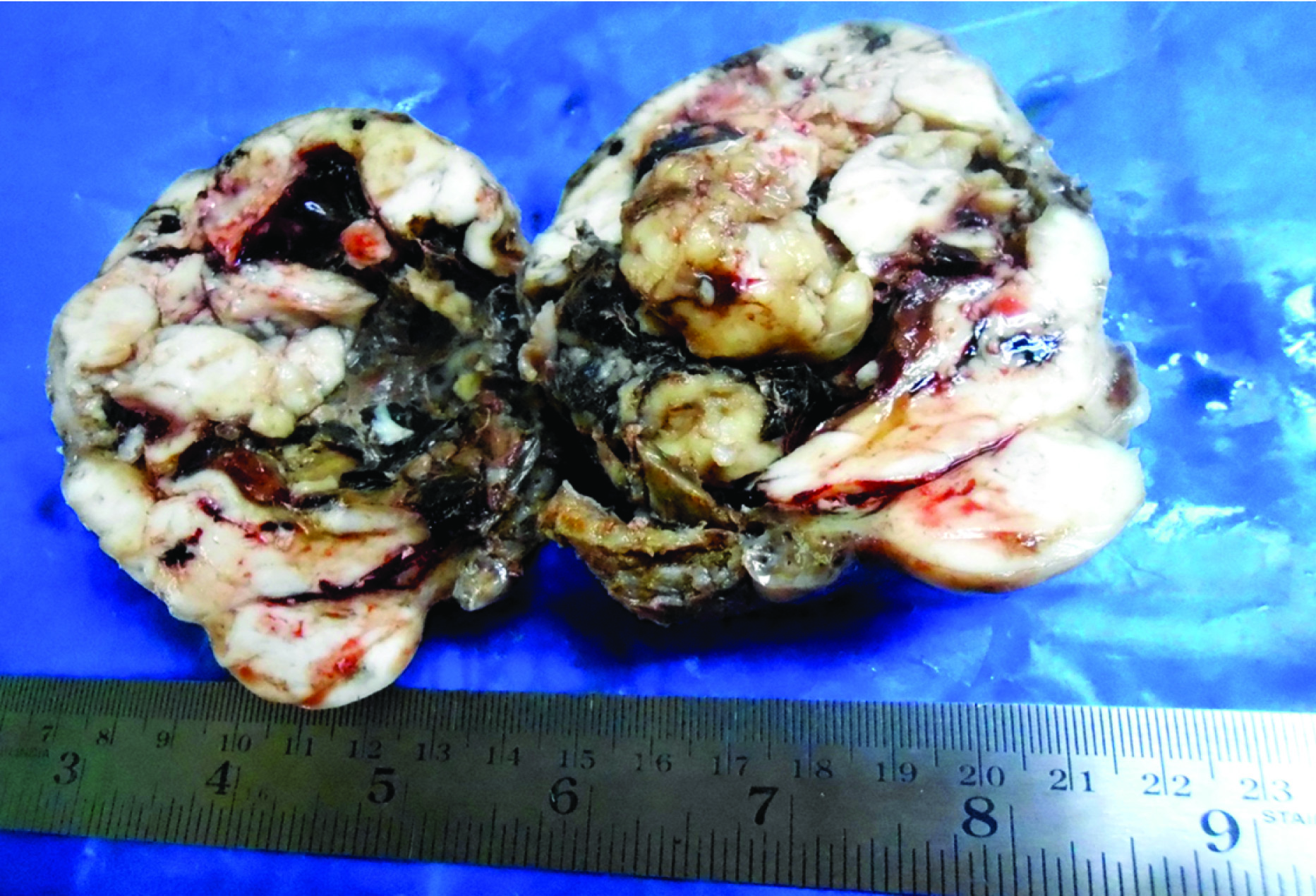
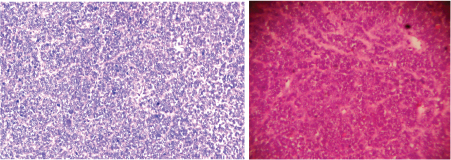
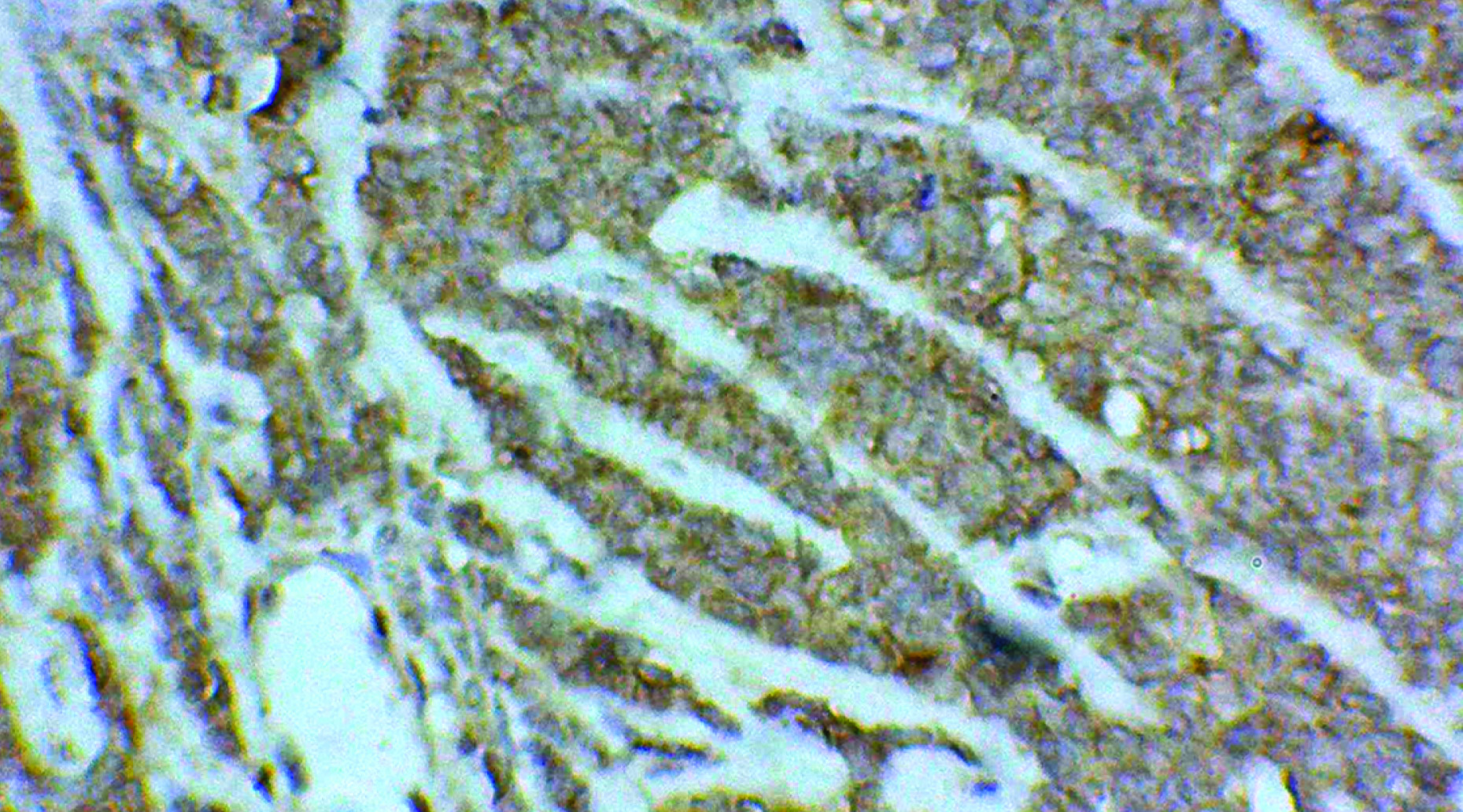
Clinically, only a palpable abdominal mass was detected other system examination was normal. Clinically it was suspected as mesenteric cyst. CT Scan revealed a huge complex sol 9.1×8.8×9.2cm with large central cystic and lobulated enhancing, solid components with foci of calcification is seem to arise from body and tail regions of pancreas extending anterior and superiorly causing displacement of gut loops. Radiological impression was large complex pancreatic tumour with cystic degeneration [Table/Fig-1]. FNAC was performed, revealed only chocolate colour fluid, cyst macrophages, and few inflammatory cells. Surgery was performed and tumour was resected and sent for histopathological examination. On gross examination of the mass, cut surface showed both solid area and cystic areas. Cystic area measuring 4x4.5 cm [Table/Fig-2] and solid area showed friability, haemorrhage and necrosis. Histopathological examination was done by routine haematoxylin and eosin stain. Microscopically, a highly cellular tumour, made up of uniform epithelial cells arranged in solid sheets and nests, admixed with well-formed acinar structures and occasional dilated ductular formations were seen [Table/Fig-3a&b,4]. Areas of necrosis were seen within the tumour. Immunohistochemistry was done with alpha fetoprotein, showed positivity [Table/Fig-5]. Typical histological features and immunohistochemistry were suggestive of the diagnosis of pancreatoblastoma.
CT Scan showing the mass measuring (9.1x8.8x9.2) cm
Gross picture of the mass showing solid, cystic and necrotic area
(a) Histological examination of mass showing epithelial cells arranged in solid sheets and nests, with acinar structures (100 x) H&E. (b) Histological examination of mass (100 x) H&E.
Microscopical examination showing the mass arising from pancreas (scanner view H&E)
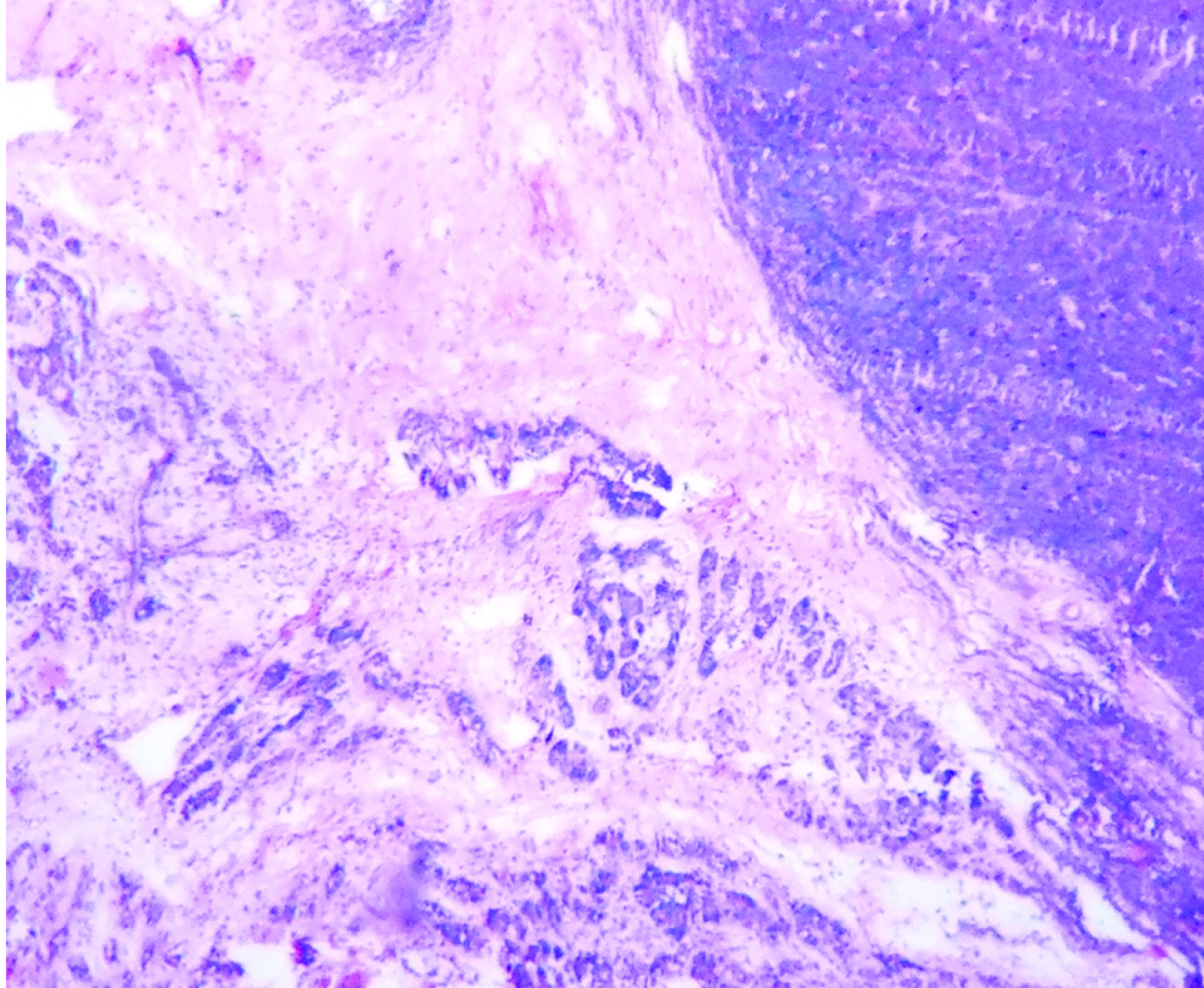
Immunohistochemistry showing positivity for alphafetoprotein (400 x)
Discussion
Pancreatoblastoma is a rare tumour; nevertheless, it is among the most common malignant neoplasms in pancreas [1]. So far <200 cases have been reported [2]. Most commonly used tumour marker is Alpha-Fetoprotein (AFP) [3]. The majority of pancreatoblastomas occur in children, and the median age of paediatric patients is approximately 4 years, and only a few cases have been described in the second decade of life. A few numbers of congenital forms were reported. The congenital form is associated with Beckwith-Wiedemann syndrome and has been described as being cystic [4]. M:F ratio is of 1.3:1. About 50% cases the head of the gland is affected, the body and the tail are in equal proportion in remainder percentage. Horie et al., divided the tumour into two categories as right-side tumours and left-side tumours [5]. According to Horie et al., the right-side tumours are usually well-encapsulated arise from the primordial ventral pancreas, do not contain calcification and islet cells, and have a better prognosis [5]. On the contrary, the left-side tumours arise from the primordial dorsal pancreas, contain calcification and islet cells, have no encapsulation, and indicate a poorer prognosis than right-side tumours [5,6]. Our patient has the features of a left-side tumour because it arises from the body of pancreas.
Histologically pancreatoblastoma shows hypercellularity with acinar differentiation and characteristic squamoid corpuscules. Large islands of plump, epithelioid cells, may be whorling nests of spindled cells or keratinizing islands are seen in squamoid corpuscles The nuclei of the squamoid corpuscles are larger and more oval than those of the surrounding cell, due to the accumulation of biotin nuclear clearing are seen [7].
Over 90% of pancreatoblastomas exhibit evidence of acinar differentiation in the form of PAS-positive, diastase resistant cytoplasmic granules as well as immunohistochemical staining for pancreatic enzymes, including trypsin, chymotrypsin, and lipase. The staining may be focal, often limited to the apical cytoplasm in areas of the tumour with acinar formations. At least focal immunoreactivity for markers of endocrine differentiation (chromogranin or synaptophysin) may be found in two third of cases.
Presence of acinar cells containing zymogen like granules on electron microscopy helps in confirmation of diagnosis. Some forms of pancreatoblastoma may have both exocrine endocrine components associated with presence of both zymogen and neuro-endocrine granules, sometimes in the same cells, or in different cells [8]. This differentiation may predict good prognosis [9]. Immunohistochemistry may be positive with alpha fetoprotein within solid regions of epithelial component, these patient have may have serum marker of elevated alpha fetoprotein.
As the presenting feature is nonspecific, diagnosis of this upper abdominal mass is rarely, made preoperatively, and other more common conditions are generally considered in differential diagnosis like neuroblastoma, hepatoblastoma, malignant lymphoma, pseudocyst and Wilms’ tumour, desmoblastic small cell tumour. Acinar cell carcinoma have similar histological and radiological feature with pancreatoblastoma. Our case was initially diagnosed by surgeon as mesenteric cyst, but after surgical excision histological examination and immunohistochemical study confirmed the diagnosis. Serum Alphafoetoprotein was estimated after suspicion and it showed higher value.
Though all three aspects of treatment may be considered, but surgery is the only means of current therapy [10]. Surgery requires complete resection of tumour, sometimes a pancreatoduodenectomy (Whipple procedure) is needed [11]. Chemotherapy, alone or in combination with radiotherapy may be considered in preoperative or palliative setting in inoperable or metastatic cases. Local radiotherapy has been proposed before or after surgery 1in some studies [11]. Camprodon and Quintanilla had given opinion that the tumour may be cured by radiotherapy only [10]. There are few reports about chemotherapy. Various combinations of chemotherapeutic regimen are used, but none of the regimen was found superior to other. As the tumour has low frequency, the evaluation of the other drugs is difficult [12].
Conclusion
A case of pancreatoblastoma is reported. As it has favourable prognosis, pancreatoblastoma should be differentiated from other aggressive tumours in children and early diagnosis is needed for specific treatments.
[1]. Perez EA, Gutierrez JC, Koniaris LG, Neville HL, Thompson WR, Sola JE, Malignant pancreatic tumours: Incidence and outcome in 58 pediatric patientsJ Pediatr Surg 2009 44:197-203. [Google Scholar]
[2]. Shorter NA, Glick RD, Klimstra DS, Brennan MF, Laquaglia MP, Malignant pancreatic tumours in childhood and adolescence: The Memorial Sloan-Kettering experience, 1967 to presentJ Pediatr Surg 2002 37(6):887-92. [Google Scholar]
[3]. Morohoshi T, Sagawa F, Mitsuya T, Pancreatoblastoma with marked elevation of serum alpha-fetoprotein. An autopsy case report with immunocytochemical studyVirchows Arch A Pathol Anat Histopathol 190 416:265-70. [Google Scholar]
[4]. Drut R, Jones MC, Congenital pancreatoblastoma in Beckwith- Wiedemann syndrome: an emerging associationPediatr Pathol 1988 8:331-39. [Google Scholar]
[5]. Horie A, Yano Y, Kotoo Y, Miwa A, Morphogenesis of pancreatoblastoma, infantile carcinoma of the pancreas: Report of two casesCancer 1977 39:247-54. [Google Scholar]
[6]. Herman TE, Siegel MJ, Dehner LP, CT of pancreatoblastoma derived from the dorsal pancreatic anlageJ Comput Assist Tomogr 1994 18:648-50. [Google Scholar]
[7]. Tanaka Y, Ijiri R, Yamanaka S, Kato K, Nishihira H, Nishi T, Pancreatoblastoma: optically clear nuclei in squamoid corpuscles are rich in biotinMod Pathol 1998 11:945-49. [Google Scholar]
[8]. Ichijima K, Akaishi K, Toyoda N, Carcinoma of the pancreas with endocrine component in childhoodAm J Clin Pathol 1985 83:95-100. [Google Scholar]
[9]. Hamoudi A, Misugi K, Grosfeld JL, Papillary\ epithelial neoplasm of pancreas in a child. Report of a case with electron microscopyCancer 1970 26:1126-34. [Google Scholar]
[10]. Camprodon R, Quintanilla E, Successful long-term results with resection of pancreatic carcinoma in children: favourable prognosis for an uncommon neoplasmSurgery 1984 95(4):420-26. [Google Scholar]
[11]. Griffin BR, Wisbeck WM, Schaller RT, Radiotherapy for locally recurrent infantile pancreatic carcinoma (pancreatoblastoma)Cancer 1987 15;60(8):1734-36. [Google Scholar]
[12]. Agrawal I, Bansal N, Bahadur AK, Rathi AK, Singh K, Pancreatoblastoma: A Case Report and Review of LiteratureInd j med & paed onco 2006 27(4):28-31. [Google Scholar]