A Case Report of Dyschromatosis Universalis Hereditaria (DUH) with Primary Ovarian Failure (POF)
N.S Jayanthi1, V. Anandan2, W. Afthab Jameela3, V. Senthil Kumar4, P. Lavanya5
1 Assistant Professor, Department of Dermatology, Stanley Medical College, Chennai, Tamil Nadu, India.
2 Professor and Head of Department, Department of Dermatology, Stanley Medical College, Chennai, Tamil Nadu, India.
3 Associate Professor, Department of Dermatology, Stanley Medical College, Chennai, Tamil Nadu, India.
4 Assistant Professor, Department of Dermatology, Stanley Medical College, Chennai, Tamil Nadu, India.
5 Resident, Department of Dermatology, Stanley Medical College, Chennai, Tamil Nadu, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. P. Lavanya, W/o Dr. M. Sivasankar, G1, Block2, Sindur Park, N0:30/31, Dr. Gurusamy Road, Chetpet, Chennai – 600031, India. E-mail : drlavanyasivasankar@gmail.com
Dyschromatosis Universalis Hereditaria (DUH) belongs to a group of congenital diffuse reticulate pigmentary disorders characterised by both hypo and hyper pigmented macules. It is both hereditary and sporadic. A number of associated cutaneous and systemic diseases have been reported. The recent discovery of the mutation in ATP binding cassette protein, ABCB6 in DUH attempts to explain the reason behind the pigmentary abnormalities and varied associations. We add a new association by reporting a case of DUH with primary ovarian failure (POF) and hypothyroidism.
ATP-binding cassette transporters, Hypothyroidism, Primary ovarian insufficiency
Case Report
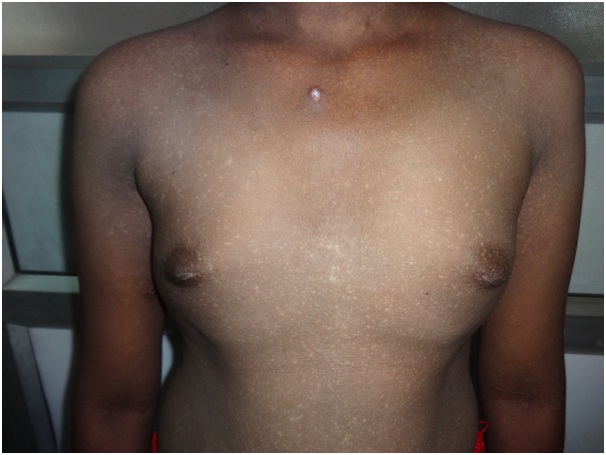
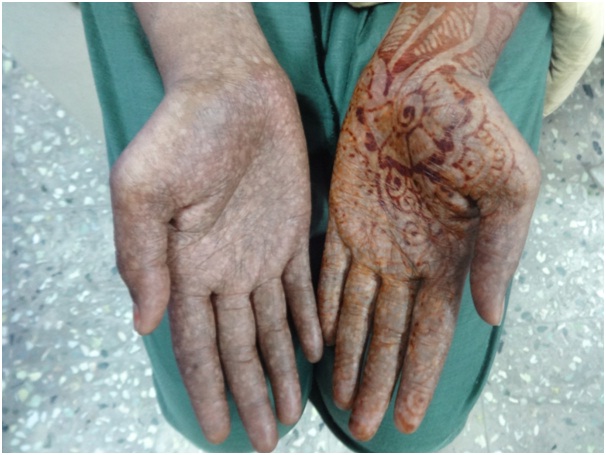
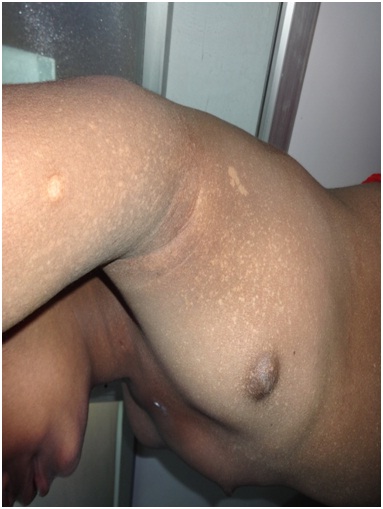
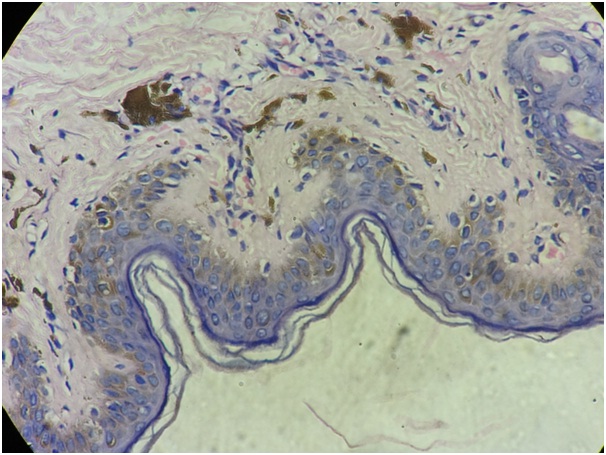
An 18-year-old female presented with history of mottled pigmentation all over the body since birth. She had not attained menarche and was on medication for hypothyroidism for the past one year. She was the first born of a second degree consanguineous marriage. There was no history of similar disease in any of her family members. On examination she was of normal stature and average build. Dermatological examination revealed multiple hypo and hyper pigmented macules in a reticulate and mottled pattern over chest, back, upper and lower extremities sparing the face [Table/Fig-1]. Palms and soles were involved [Table/Fig-2]. Dermatoglyphic pattern was preserved. Scalp, hair, nails and mucosae were normal. Secondary sexual characters were poorly developed and corresponded to Tanner Stage 2 [Table/Fig-3]. No signs of virilisation were observed. On investigation, karyotyping was 46XX [Table/Fig-4]. Blood estradiol level was pre-pubertal (5pg/ml against a normal of 20 – 200pg/ml) and FSH level was elevated (130.20 mIU/ml against a normal of 3.5- 12.5mIU/ml - Hypergonadotropic hypogonadism). Serum testosterone level was also elevated (17.83pg/ml against a normal of 0.02 – 3.09pg/ml). Sex hormone binding globulin level was sub-normal (23.39nmol/L against a normal of 26.1 – 110nmol/L). Serum LH (39.69mIU/ml; normal value: 14.0 – 95.6mIU/ml) and prolactin levels (12.26ng/ml; normal value: 4.79 – 23.3ng/ml) were normal. Serum T3 (3.22pg/ml; normal value: 2.3 – 4.2pg/ml) and T4 levels (1.14ng/dl; normal value: 0.8 – 2.0ng/dl) were normal with medications and TSH level was slightly elevated (7.560mIU/ml; normal value: 0.35 -5.50mIU/ml). Random blood sugar was normal (96mg/dl) but fasting insulin level was slightly elevated (27.76mIU/ml against a normal value of 2.6 – 24.9mIU/ml). An ultrasonogram showed prepubertal uterus with tubular configuration and bilateral small ovaries with few follicles. Complete blood count, liver and renal function tests, anti-thyroid antibody levels and anti-nuclear antibody titre were normal. Hearing and vision was normal. Cardiovascular, respiratory, and central nervous system examination revealed no abnormality clinically. Her IQ was normal. Skin biopsy showed normal epidermis with increased pigmentation of basal layer with melanin incontinence in the dermis [Table/Fig-5]. A diagnosis of Dyschromatosis Universalis Hereditaria (DUH) with Primary Ovarian Failure (POF) and hypothyroidism was made.
Multiple hypo pigmented macules in a reticulate and mottled pattern overchest.
Poorly developed secondary sexual characters.

High power view of histopathology shows increased pigmentation of basal layer and pigment incontinence in the dermis.
Patient was started on cyclical hormonal supplements for development of secondary sexual characters and prevention of osteoporosis after consultation with an endocrinologist and gynaecologist. Genetic counselling was given.
Discussion
DUH was first described in Japan by Ichikawa and Hiraga in 1933 [1]. The disease was initially thought to be confined to Japan but several case reports have been made from Europe, China, Saudi Arabia, Tunisia and India [2]. Autosomal dominant, autosomal recessive and sporadic cases have been documented. Another similar disorder, Dyschromatosis Symmetrica Herediteria (DSH) was initially thought to be a localised form of DUH. But DSH had mutations in ADAR1 gene, localised to chromosome 1q21.3 and this mutation was not present in DUH [3]. Now they are considered as two different entities. DSH involves the face and dorsal extremities whereas DUH involves mainly the trunk with or without sparing of palms, soles and face [4]. However, palmar and oral mucosal involvement [5], nail and hair involvement [6] and adermatoglyphia [7] have been documented.
DUH was thought to be a disorder of melanocyte number, melanosome production or distribution of epidermal melanin unit [8]. Recently the molecular basis of DUH was identified by a genome wide linkage analysis in a Chinese pedigree and confirmed using DNA sequencing and animal models [9]. The candidate gene was identified as ABCB6 gene located on chromosome 2q36 and is a member of ATP Binding Cassette (ABC) transporters. ABC transporters, responsible for transport of various molecules across cell membranes, are present ubiquitously including the skin. They play a role in cellular transition and metal homeostasis, especially copper. It is thought that mutations of ABCB6 leads to abnormalities in copper homeostasis thereby causes dysfunction of tyrosinase and further leads to abnormal melanin synthesis. It is also expressed in exosomes and plays a role in melanosome transport to surrounding keratinocytes [10].
ABCB6 protein has different functions at different sites and hence mutations of ABCB6 domains produces disease with diverse phenotypes. Consistently, DUH has been reported to be associated with a wide range of cutaneous and systemic diseases such as tuberous sclerosis, learning difficulties, mental retardation [4], short stature and high tone deafness [11], X- Linked oculo-cutaneous albinism [12], epilepsy [13], erythrocyte, platelet and tryptophan metabolism abnormalities [14] and renal failure [15].
Regarding the cause of POF, no precipitating factors like viral infections, irradiation or chemotherapy in childhood could be identified. Her karyotyping was normal. There was no evidence of adrenal insufficiency. Antithyroid antibody profile was normal. As antiovarian antibodies are neither sensitive nor specific in diagnosing autoimmune oophoritis as a cause of POF, it was not measured. FSH receptor polymorphisms also can cause hypergonadotropic hypogonadism. Interestingly, the FSH receptor is also localised to chromosome 2 where the gene for ABCB6 protein is located. However, in more than 90% of patients with POF, no cause could be identified.
Possible differential diagnoses for skin lesion include xeroderma pigmentosum, dyskeratosis congenita and other reticulate pigmentary disorders. The absence of photosensitivity, presence of skin lesions in covered areas of the body, asymptomatic nature, absence of nail, mucosal and scalp involvement rules out the above disorders. There is no specific treatment for the skin condition. Removal of the skin lesions with Q-switched alexandrite laser has been tried [16]. DUH with POF has not been reported in literature so far to the best of our knowledge.
Conclusion
We report this case for its rarity and to highlight the recent discovery of the molecular pathogenesis behind DUH and its diverse systemic associations. Advanced genetic analysis may elucidate the association of DUH with POF taking into consideration the co-localization of the FSH receptor gene and ABCB6 gene on chromosome 2.
[1]. Ichigawa T, Hiraga Y, A previously undescribed anomaly of pigmentation dyschromatosisuniversalishereditariaJpn J Dermatol Urol 1933 34:360-64.(in Japanese) [Google Scholar]
[2]. Sethuraman G, D’Souza M, Thappa DM, Srinivas CR, Smiles L, Dyschromatosis universalis hereditariaClin Exp Dermatol 2002 27(6):477-79. [Google Scholar]
[3]. Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, Mutation Analysis of the ADAR1 Gene in Dyschromatosis Symmetrica Hereditaria and Genetic Differentiation from both Dyschromatosis Universalis Hereditaria and Acropigmentatio ReticularisJ Invest Dermatol 2005 124(6):1186-92. [Google Scholar]
[4]. Irvine AD, Mellerio JD, Genetics and genodermatoses. In: Burns T, Breathnach S, Cox N, Griffiths C. EdsRook’s textbook of Dermatology 2010 8th edOxfordBlackwell Science:445-598. [Google Scholar]
[5]. Binitha M, Thomas D, Asha L, Tuberous sclerosis complex associated with dyschromatosisuniversalishereditariaIndian J Dermatol Venereol Leprol 2006 72(4):300 [Google Scholar]
[6]. Naik CL, Singh G, Rajashekar TS, Okade R, Dyschromatosis universalis hereditariaIndian J Dermatol 2009 54(5):74 [Google Scholar]
[7]. Bhoyar P, Mahajan B, Kumar S, A case of dyschromatosisuniversalishereditaria with adermatoglyphia: A rare associationIndian Dermatol Online J 2015 6(2):105 [Google Scholar]
[8]. Nuber UA, Tinschert S, Mundlos S, Hauber I, Dyschromatosis universalis hereditaria: Familial case and ultrastructural skin investigationAm J Med Genet A 2004 125A(3):261-66. [Google Scholar]
[9]. Liu H, Li Y, Hung KKH, Wang N, Wang C, Chen X, Genome-Wide Linkage, Exome Sequencing and Functional Analyses Identify ABCB6 as the Pathogenic Gene of Dyschromatosis Universalis HereditariaPLoS ONE 2014 9(2):e87250 [Google Scholar]
[10]. Zhang C, Li D, Zhang J, Chen X, Huang M, Archacki S, Mutations in ABCB6 cause dyschromatosis universalis hereditariaJ Invest Dermatol 2013 133(9):2221-28. [Google Scholar]
[11]. Rycroft RJG, Calnan CD, Wells RS, Universal dyschromatosis, small stature and high-tone deafnessClin Exp Dermatol 1977 2(1):45-48. [Google Scholar]
[12]. Yang JH, Wong CK, Dyschromatosis universalis with X-linked ocular albinismClin Exp Dermatol 1991 16(6):436-40. [Google Scholar]
[13]. Pavithran K, Dyschromatosis Universalis Hereditaria with EpilepsyIndian J Dermatol Venereol Leprol 1991 57(2):102 [Google Scholar]
[14]. Pirasath S, Sundaresan T, Tamilvannan T, Thrombocytopenia in dyschromatosis universalis hereditariaCeylon Med J 2012 57(3):124-24.[Internet] [cited 2016 Jan 3] Available from: http://www.sljol.info/index.php/CMJ/article/view/4705 [Google Scholar]
[15]. Rojhirunsakool S, Vachiramon V, DyschromatosisUniversalisHereditaria with Renal FailureCase Rep Dermatol 2015 7(1):51-55. [Google Scholar]
[16]. Nogita T, Mitsuhashi Y, Takeo C, Tsuboi R, Removal of facial and labial lentigines in dyschromatosis universalis hereditaria with a Q-switched alexandrite laserJ Am Acad Dermatol 2011 65(2):e61-63. [Google Scholar]