Angiosarcoma of the breast is an unusual tumour. The diagnosis of this condition is difficult clinically, pathologically and radiologically. Treatment includes surgery followed by chemoradiation. The tumour is notorious for its recurrence and is generally associated with poor prognosis. Kasabach-Merritt syndrome also is a very rare entity seen primarily in infants with vascular malformations and carries a poor prognosis. In this report, the authors describe a patient with recurrent primary angiosarcoma of the breast, which presented as Kasabach-Merritt syndrome.
Case Report
A 38-year-old lady presented to the surgical out-patient department with insidious onset of multiple reddish lumps in her right breast since 8 months which were progressively enlarging. There was no pain associated with the lesion. There was no history of fever. There was no history of other malignancies, trauma or radiation to the chest wall. She was married with two children who were breast-fed up to 9 months of age. There was no family history breast malignancy. The contralateral breast and bilateral axillae were unremarkable. Clinically, the lesion measured 10x12cm in size and was diagnosed as malignant breast tumour. Ultrasound of the lesion showed a heterogeneous mass with increased vascularity and containing both hyperechoic and hypoechoic components. Fine needle aspiration cytology showed a lesion suspicious of Angiosarcoma, which was confirmed after performing a core needle biopsy [Table/Fig-1]. The patient underwent a right-sided simple mastectomy. Confirmation of the pre-operative diagnosis was obtained following histopathological examination and immunohistochemistry of the resected specimen [Table/Fig-2,3 and 4]. Following surgery, the patient did not consent for adjuvant chemoradiotherapy and was hence discharged. The patient came back to the casualty 4 months later with complaints of severe weakness. On examination, she appeared pale and it seemed like the tumour had recurred at the scar site [Table/Fig-5]. Her blood picture showed anaemia, thrombocytopaenia, hypofibrinogenaemia, prolonged prothrombin and activated prothrombin time, increased levels of fibrinogen degradation products and d-dimers. The patient was diagnosed to have tumour recurrence and a Kasabach-Merritt Syndrome-like phenomenon. Patient was managed conservatively by transfusion of packed cells, platelets and fresh frozen plasma. Contrast enhanced computed tomography of the chest revealed an ulcerated, heterogeneous, minimally enhancing soft tissue lesion with loss of fat plane between the lesion and the underlying pectoralis major muscle [Table/Fig-6]. Work-up for metastasis was negative. The patient was taken up for a wide local excision of the tumour [Table/Fig-7,8]. After surgery, her platelet count gradually increased and there was a drop in the levels of fibrinogen degradation products and d-dimers. Currently, the patient has been started on adjuvant Paclitaxel and radiotherapy has been planned.
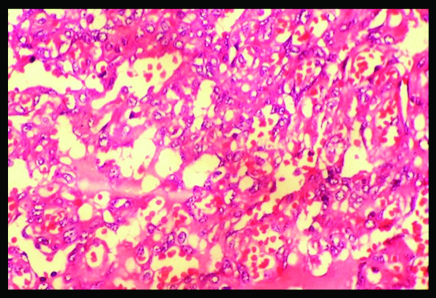
Core Needle Biopsy of the lesion, 40x showing features suggestive of Angiosarcoma.
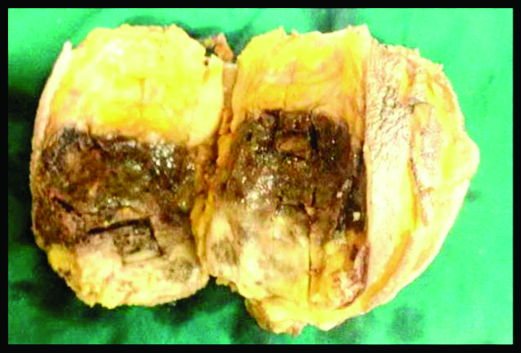
Gross simple mastectomy specimen with grey-brown tumour showing haemorrhaging areas.
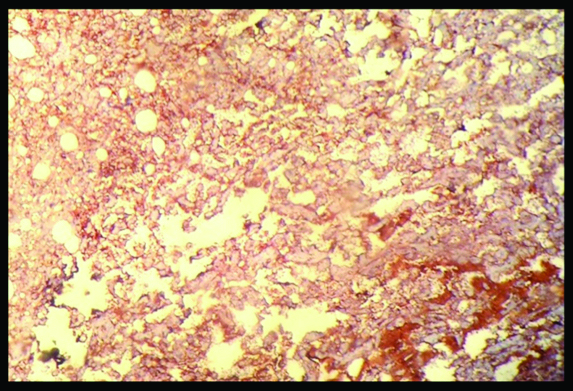
Immunohistochemistry positive for Factor VIII (40x).
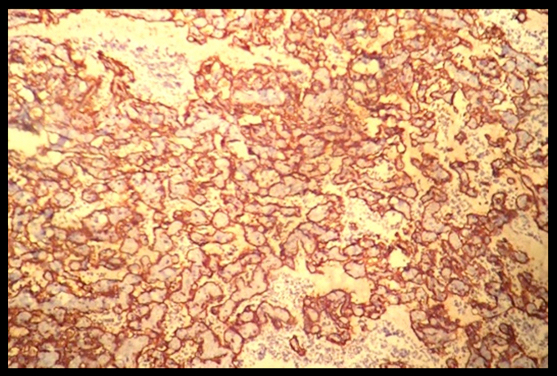
Immunohistochemistry positive for CD 34 (40x).
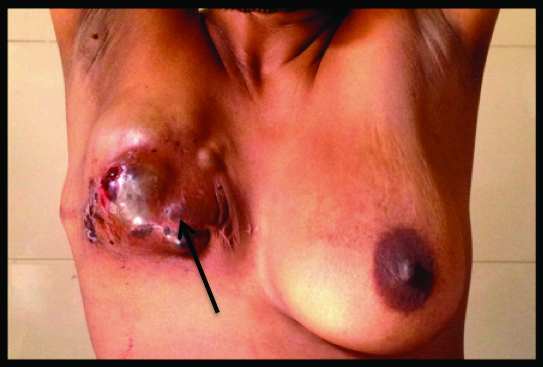
Patient presented with recurrence at scar site (indicated by black arrow).
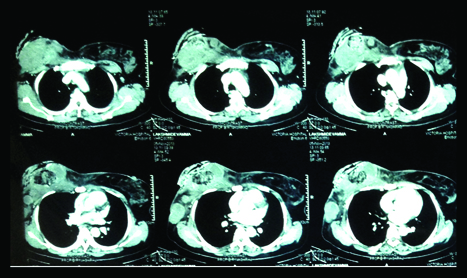
Contrast CT revealed an ulcerated, heterogenous, minimally enhancing soft tissue lesion with loss of fat plane between the lesion and the underlying pectoralis major muscle.
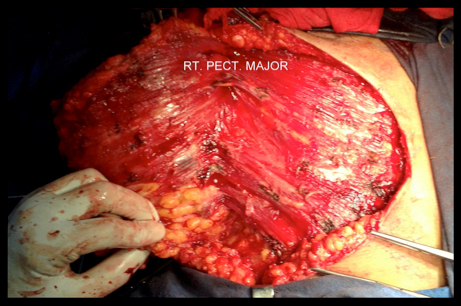
Intraoperative image of recurrent tumour being excised.
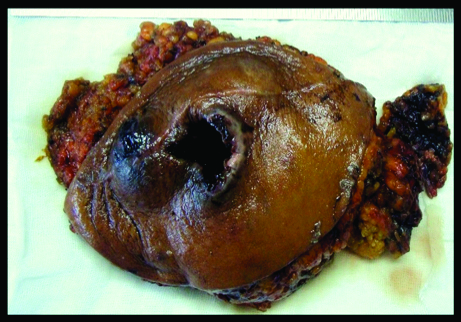
Gross image of the recurrent lesion.
Discussion
Angiosarcomas account for less than 1% of all sarcomas making them the rarest in that category. Enzinger and Weiss have classified them into four groups: (a) Cutaneous Angiosarcoma associated with lymphedema; (b) Cutaneous Angiosarcoma not associated with lymphedema; (c) Angiosarcoma of the breast; and (d) Angiosarcoma of deep soft tissue [1]. Schmidt described Angiosarcoma (AS) of the breast in 1887 [2]. These are rare breast tumours. Of these, only 20% are primary and represent 0.04% of all malignant breast tumours with an annual incidence of primary breast angiosarcoma estimated to be about 5.8 new cases per ten million women [3–7]. These are however the most common type of sarcomas arising in the breast [8]. Broadly these tumours are classified into primary and secondary, the difference being that secondary angiosarcomas are tumours that arise in patients who have undergone previous breast surgeries, irradiation or have lymphoedema. Some authors also claim trauma and breast implants to be risk factors for the development of angiosarcomas [9].
Clinically, primary angiosarcomas are seen in younger women with a median age of onset between 20 and 40 years whereas secondary angiosarcomas are reported approximately 12.5 years following radical mastectomy and about 10 years following lymphedema [1,4]. Around 6 to 12% of cases of primary angiosarcomas of the breast are diagnosed peri-partum which suggests probable hormonal involvement in causation of the disease. But cases with proven oestrogen-receptor involvement are so infrequent that is not possible to link hormonal involvement and angiosarcomas [3,10]. Patients with primary angiosarcomas generally present with painless fast growing discrete palpable masses that have insidious clinical onset [8]. Some may complain of pain associated with the lump and approximately 2% of the patients can present with diffuse enlargement of the breast. There is usually no nipple retraction, discharge or axillary nodes enlargement. A rare case of this tumour occurring in a postmenopausal woman and bilateral breast involvement has been reported [10]. Interestingly, at the time of presentation most tumours have measured more than 4 cm according to the available literature [3].
The diagnosis of AS can be delayed due to unclear imaging findings. Mammography may reveal skin thickening and ill-defined superficial mass. Because these patients are often young and have dense breasts, mammograms may be normal in up to one-third of cases [5,9,11,12]. Rarely, the tumour may show a soap bubble appearance. They may occasionally present as coarse non-branching microcalcifications [13]. On ultrasound, angiosarcomas present as a hypervascular, heterogeneous and hyperechoic mass with architectural distortion [4,11]. There may be multiple interfaces of vascular channels and this might alert the radiologist to a diagnosis of angiosarcoma [7]. An MRI of angiosarcoma shows a heterogeneous mass with low signal intensity on T1-weighted images, but signal intensity is high in images that are T2-weighted. The latter suggests the presence of vascular channels containing slow flowing blood. The tumour may appear as a rapidly enhancing heterogenous mass with haemorrhage and blood lakes [4,13]. Although not definitive, MRI is useful in ascertaining the extent of tumour and in planning surgery [9]. Dynamic study using contrast medium could show prolonged enhancement that may reflect the histological features including blood-filled vascular spaces and vascular channels [14]. Positron emission tomography with 18F-fluorodeoxyglucose (FDG-PET) may be used for staging of angiosarcoma [3,9].
Histologically, diagnosis by fine needle aspiration cytology (FNAC) or core needle biopsy (CNB) is difficult. A false negativity rate of 37% has been reported after percutaneous biopsy was done. Large-core biopsies might facilitate the correct diagnosis as they provide a larger sample, but such a macrobiopsy is often difficult to perform due to the vascular nature of these tumours. Surgical resection and microscopic examination of sufficient sample of the tumour are often necessary to render a final diagnosis [13].
Angiosarcomas of the breast are classified into 3 grades (grade 1–3) or as well to poorly differentiated tumours by Donnel et al., [15]
Grade I (well differentiated): These contain open anastomosing vascular channels that proliferate within dermis, subcutaneous tissue or breast tissue. A single layer of endothelial cells lines these channels which dissect through the stroma causing distortion but little destruction of the preexisting lobules and ducts. The endothelial cells are usually flat; the nuclei may be hyperchromatic and contain small nucleoli. Solid and spindle cell foci, blood lakes and necrosis are not present.
Grade II (Intermediate-grade): These tumours differ from low-grade by containing additional cellular foci of papillary formations and/or solid and spindle cell proliferation. The greater part of the tumour, however, is still composed of low-grade histology. Slightly increased mitotic activity is observed.
Grade III (poorly differentiated): They have endothelial tufting and papillary formations are prominent. Conspicuous solid and spindle cell areas, mostly devoid of vascular formations, are present as well. Mitoses may be brisk, especially in more cellular areas. Areas of haemorrhage, known as ‘blood lakes’ and necrosis are also seen [8].
Correlation between grade of the tumour and prognosis has been established. Disease-free survival 5 years after initial treatment according to Rosen’s study is 76% with grade 1 tumours, up to 70% for grade 2 tumours and falls to 15% with grade 3 tumours. Disease recurrence is more than 15 years with grade 1 tumours, but in patients with grade 3 tumours, the time to recurrence averages 15 months [9,13]. However, a recent report suggests that tumour grade might not be a significant prognostic factor [16].
Differential diagnosis of this rare malignancy includes:
Benign haemangioma
Cystosarcoma phyllodes
Stromal sarcoma
Metaplastic carcinoma
Squamous cell carcinoma with sarcomatoid features
Myoepithelioma
Fibromatosis
Fibrosarcoma
Liposarcoma
High-grade mammary carcinoma especially in small biopsy specimens containing only solid areas
Reactive spindle cell proliferative lesions.
Immunohistochemistry can show positivity for factor VIII antigen, CD34, CD31, desmin and vimentin. CD31 appears to be the most promising marker [2–4,13,17].
The treatment of AS will require a multi-disciplinary team. Although there are no standard guidelines regarding treatment, surgery is usually the primary and the most common mode of management to achieve local control of this aggressive tumour. Obtaining negative margins forms the goal of surgery. The most common surgery performed is total mastectomy without axillary dissection. In cases where axillary node involvement is suspected, axillary dissection can be performed [4,17]. Recurrence rates as high as 46.5% occurring about 9 months following surgical treatment have been reported and there is no role for breast conservation [3,11]. There may be a limited role of embolization in the management of this disease [18]. There is data to suggest that angiosarcomas appear to be sensitive to chemotherapy. The tumour appears to be sensitive to taxanes and liposomal doxorubicin. Weekly Paclitaxel or liposomal doxorubicin has achieved results better than the previously used regimen involving anthracyclines and ifosfamide [4]. There is literature supporting the use of cisplatin and thalidomide as well [11]. Newer agents targeting angiogenesis such as Bevacizumab, Rapamycin and Sorafenib are being evaluated although the response rates are not convincing at this point [13,17]. The tumour cells of AS have a high growth rate. Hence hyperfractionated radiotherapy is preferred for locoregional control of AS as use of multiple daily fractions might prevent repopulation from occurring [4]. However, it has to be borne in mind that no survival benefit from radiotherapy has been proved and it is used only to reduce local recurrence [17]. Radiotherapy can also be used as palliation for bony metastases [3].
It should not be forgotten that angiosarcoma is an extremely aggressive malignancy and carries a worse prognosis than breast carcinomas [4]. The disease is frequently known to metastasize to liver followed by lung, lymph nodes, bones, bone marrow and less frequently to ovary, kidney, heart, ovary, omentum, adrenal gland, stomach, pancreas, peritoneum, esophagus, and skin [2,3].
Our patient, along with having tumour recurrence also presented with features suggestive of Kasabach–Merritt Syndrome (KMS). It is a rare disorder defined as a consumptive thrombocytopaenia in the presence of a highly vascular and locally aggressive tumour. It is usually associated with the following features:
Rapidly enlarging vascular anomaly
Consumptive coagulopathy
Thrombocytopaenia
Prolonged prothrombin time and activated prothrombin time
Hypofibrinogenaemia
Presence of d-dimer and fibrin split products with or without microangiopathic haemolytic anaemia.
KMS was first noted by Kasabach and Merritt in 1940 [19]. It was originally described in association with large benign haemangiomas in infants. It’s pathogenesis is related to platelets being trapped in highly vascular tumours with secondary consumption of clotting factors and resultant bleeding [20]. It is almost always described in children with only a handful of cases been described in adults [21]. Angiosarcoma associated with KMS is unusual is associated with a mortality rate of about 20% to 40%. Fatality in these patients occurs due to haemorrhage, cardiovascular insufficiency or infection [21]. Management of KMS includes high dose corticosteroids and interferon alpha, which work as anti-angiogenic agents. Other therapeutic modalities include compression of the lesion, arterial embolization with bleomycin, laser therapy, sclerotherapy, radiotherapy and chemotherapy with vincristine, doxorubicin, paclitaxel and use of drugs such as dipyridamole, pentoxyfylline and propranolol [19,21–23]. It should be noted that there is however no clear consensus regarding the management of KMS [22].
After going through the available literature, the authors would like to emphasize that this is the first time a diagnosis of KMS has been made in a patient with a recurrent primary angiosarcoma of the breast.
Conclusion
Both Angiosarcoma of breast and KMS are rare. Knowledge of these conditions is necessary as both of them are extremely aggressive. The untrained mind of a treating surgeon might be surprised in the rare case that both the entities seen in the same patient like in our case. Due to the propensity for very high mortality in these diseases, multi-disciplinary care is absolutely essential for management of such patients.
[1]. Yamada S, Yamada Y, Kobayashi M, Hino R, Nawata A, Noguchi H, Post-mastectomy benign lymphangioendothelioma of the skin following chronic lymphedema for breast carcinoma: a teaching case mimicking low-grade angiosarcoma and masquerading as Stewart-Treves syndromeDiagnostic Pathology 2014 9:197 [Google Scholar]
[2]. Rohan VS, Hanji AM, Patel JJ, Tankshali RA, Primary angiosarcoma of the breast in a postmenopausal patientJ Cancer Res Ther 2010 6(1):120-22. [Google Scholar]
[3]. Desbiens C, Hogue JC, L’evesque Y, Primary Breast Angiosarcoma: Avoiding a Common TrapCase Rep Oncol Med 2011 2011:517047 [Google Scholar]
[4]. Zemanova M, Machalekova K, Sandorova M, Boljesikova E, Skultetyova M, Svec J, Clinical management of secondary angiosarcoma after breast conservation therapyRep Pract Oncol Radiother 2013 19(1):37-46. [Google Scholar]
[5]. Alvarado-Miranda A, Bacon-Fonseca L, Ulises Lara-Medina F, Maldonado-Martínez H, Arce-Salinas C, Thalidomide combined with neoadjuvant chemotherapy in angiosarcoma of the breast with complete pathologic response: case report and review of literatureBreast Care 2013 8(1):74-76. [Google Scholar]
[6]. Keshav P, Hegde SS, Bilateral primary angiosarcoma of the breastCase Rep Surg 2013 2013:139276 [Google Scholar]
[7]. Lai MH, Lui CY, Mammary angiosarcoma in two patients at either end of the age spectrumHong Kong Med J 2010 16(2):141-44. [Google Scholar]
[8]. Bennani A, Chbani L, Lamchahab M, Wahbi M, Alaoui FF, Badioui I, Primary angiosarcoma of the breast: a case reportDiagn Pathol 2013 8:66 [Google Scholar]
[9]. Cao Y, Panos L, Graham RL, Parker TH 3rd, Mennel R, Primary cutaneous angiosarcoma of the breast after breast traumaProc (BaylUniv Med Cent) 2012 25(1):70-72. [Google Scholar]
[10]. Roy S, Devleena Maji T, Choudhuri P, Biswas J, Lahiri D, Bilateral angiosarcoma of breast in a young ladyIndian J Med Paediatr Oncol 2011 32(3):160-61. [Google Scholar]
[11]. Bhosale SJ, Kshirsagar AY, Patil MV, Wader JV, Nangare N, Patil PP, Primary angiosarcoma of breast: A case reportInt J Surg Case Rep 2013 4(4):362-64. [Google Scholar]
[12]. Babarovic E, Zamolo G, Mustac E, Strcic M, High grade angiosarcoma arising in fibroadenomaDiagn Pathol 2011 6:125 [Google Scholar]
[13]. Smoll NR, Farhadieh RD, Ferguson R, Findlay MW, Hunter-Smith DJ, High-grade Angiosarcoma Associated with Ruptured Breast ImplantsPlast Reconstr Surg Glob Open 2013 1(1) [Google Scholar]
[14]. Kikawa Y, Konishi Y, Nakamoto Y, Harada T, Takeo M, Ogata M, Angiosarcoma of the breast - specific findings of MRIBreast Cancer 2006 13(4):369-73. [Google Scholar]
[15]. Donnell RM, Rosen PP, Liberman PH, Angiosarcoma and othervascular tumours of the breastAm J Surg Pathol 1981 5:629-42. [Google Scholar]
[16]. Nascimento AF, Raut CP, Fletcher CD, Primary angiosarcoma of the breast: clinicopathologic analysis of 49 cases, suggesting that grade is not prognosticAm J SurgPathol 2008 32(12):1896-904. [Google Scholar]
[17]. D’Angelo SP, Antonescu CR, Kuk D, Qin L, Moraco N, Carvajal RC, High-risk features in radiation-associated breast angiosarcomasBr J Cancer 2013 109(9):2340-46. [Google Scholar]
[18]. Taib N, Yip Ch, Ranganathan S, Moosa F, Mun K, Haemorrhaging lesion in the breast: is there a role for embolisation?Biomed Imaging Interv J 2006 2(3):e30 [Google Scholar]
[19]. Acharya S, Pillai K, Francis A, Criton S, Parvathi VK, Kasabachmerritt syndrome: management with interferonIndian J Dermatol 2010 55(3):281-83. [Google Scholar]
[20]. Massarweh S, Munis A, Karabakhtsian R, Romond E, Moss J, Metastatic angiosarcoma and kasabach-merritt syndromeRare Tumours 2014 6(2):5366 [Google Scholar]
[21]. Tan SM, Tay YK, Liu TT, Mancer K, Cutaneous angiosarcoma associated with the Kasabach-Merritt syndromeAnn Acad Med Singapore 2010 39(12):941-42. [Google Scholar]
[22]. Wang P, Zhou W, Tao L, Zhao N, Chen XW, Clinical analysis of Kasabach-Merritt syndrome in 17 neonatesBMC Pediatr 2014 14:146 [Google Scholar]
[23]. Arunachalam P, Kumar VR, Swathi D, Kasabach-Merritt syndrome with large cutaneous vascular tumoursJ Indian Assoc Pediatr Surg 2012 17(1):33-36. [Google Scholar]