As a rare multisystem congenital anomaly disorder, Cornelia de Lange syndrome (CdLS) is featured by delayed growth and development, distinct facial dimorphism, limb malformations and multiple organ defects. CdLS is a genetic syndrome affecting 1/10000-1/60000 neonates with unknown genetic basis. Delayed growth and development, hirsute, structural anomalies of the limbs and distinct facial dimorphism are considered as its main clinical characteristics. Introducing CdLS cases of different ethnic backgrounds could add distinctions to the phenotypic picture of the syndrome and be useful in diagnosis. Early diagnosis and decreased death rates are achievable through enhanced awareness on this syndrome. We present here a 45-day-old girl, as the first case of Cornelia in Golestan (Northern Iran), referred to our hospital with the symptoms as mentioned above.
Case Report
The patient was a 45-day-old incognito girl presented, from an orphanage, to our hospital (Taleghani Children’ Hospital, Gorgan, Iran) by a caretaker with the chief complaint of productive coughs and rhinorrhea. She was born low birth weight of preterm labour through natural vaginal delivery; her family identification and history was unknown. After admission in NICU at 3 days of age, respiratory support with mechanical ventilation was performed for her and she was discharged with stable general condition.
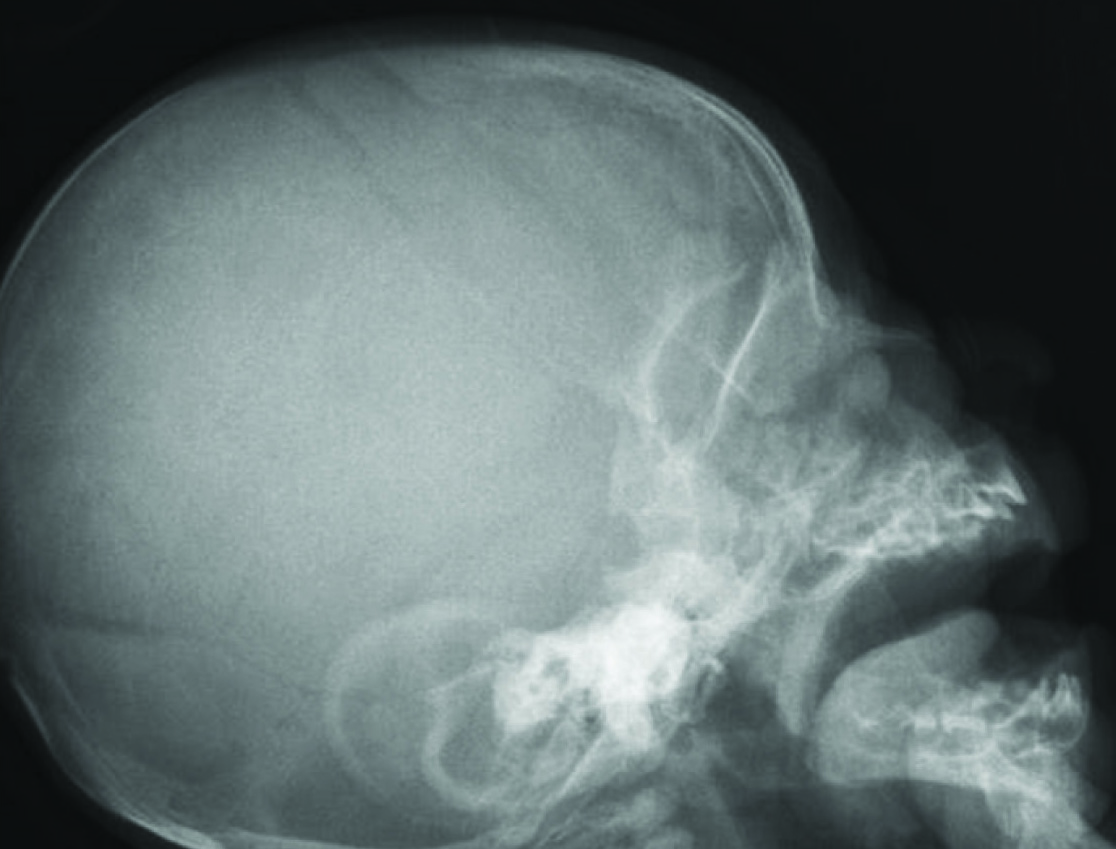
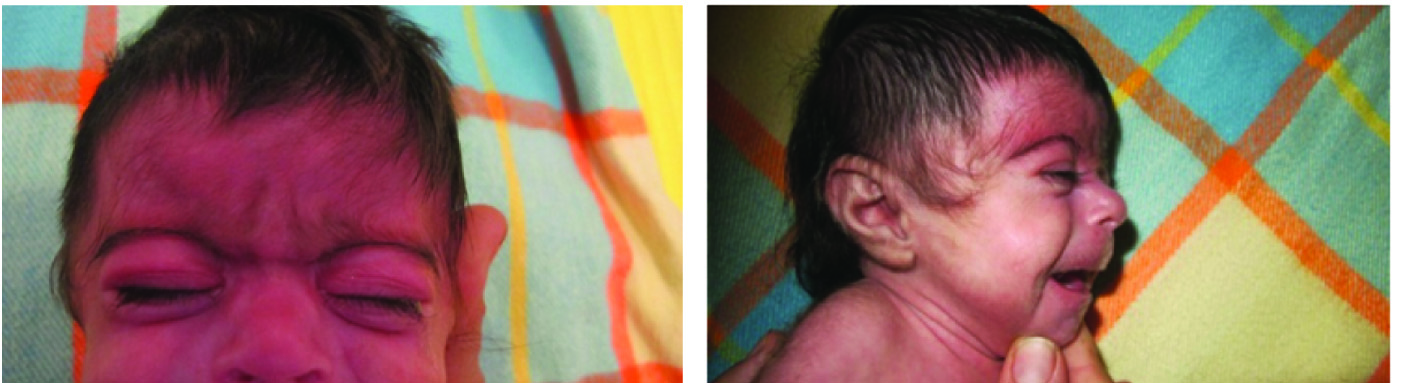
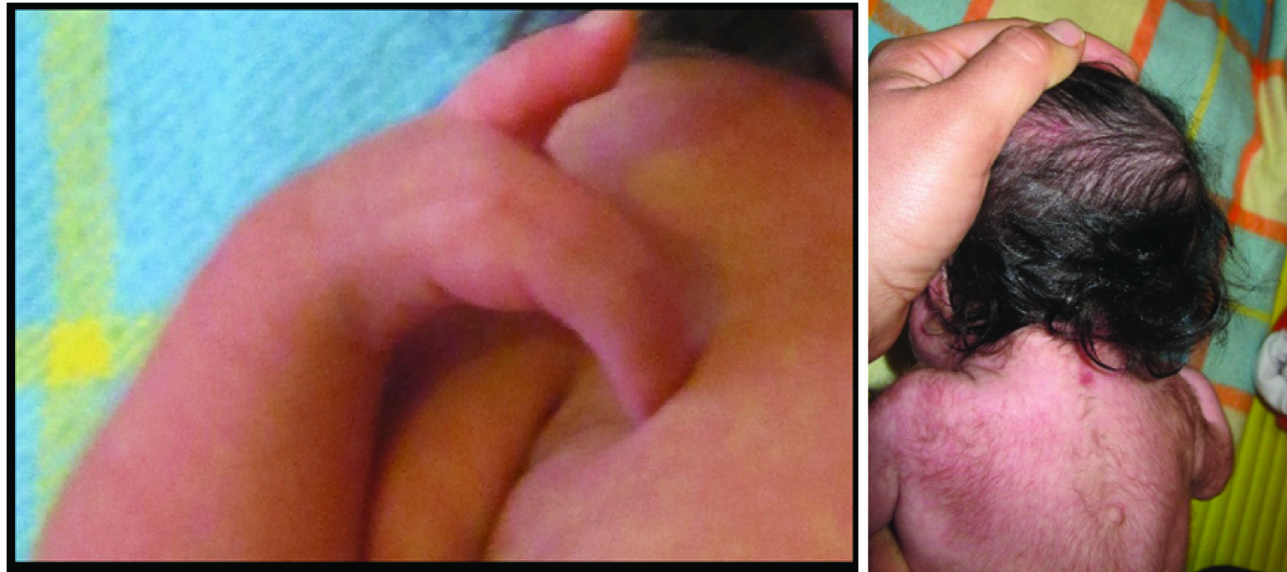
Her physical examination revealed confluent eyebrow well-defined and arch liked, long curly eyelashes, low front and back hairline, depressed nasal bridge, turned up nose, high arched palate [Table/Fig-1], down-turned angles of the mouth and thin lip, short neck, low anterior and posterior hairline, microcephaly [Table/Fig-2], excessive body hair, phocomelia [Table/Fig-3] and small broad hands with simian crease, short leg, hyper tonicity, and small labia major. A diaper rash was also evident on the skin of buttock area.
Radiograph of the lateral skull focusing high arched palate.
Head and neck appearance of the case showing her: a) confluent eyebrows; b) depressed nasal bridge, turned up nose, hirsutism, down-turned angles of the mouth and thin lip, short neck, low anterior and posterior hairline.
a) Phocomelia presented in the right hand; b) excessive body hair (hirsutism).
Her weight was 2kg and she was 41cm tall. Her head circumference was 31cm exact birth weight data was not available. On Cardiac auscultation, ejection systolic soufflé of 2/6 in pulmonary are was heard. Her trans-thoracic echocardiography revealed “valvular pulmonary stenosis” (PS), “perforated forman ovale” (PFO) as well as “partial anomalous pulmonary venous drainage” (PAPVD). Ophthalmologic examinations were unremarkable.
Laboratory analysis including complete blood count, biochemical parameters and urinalysis were normal.
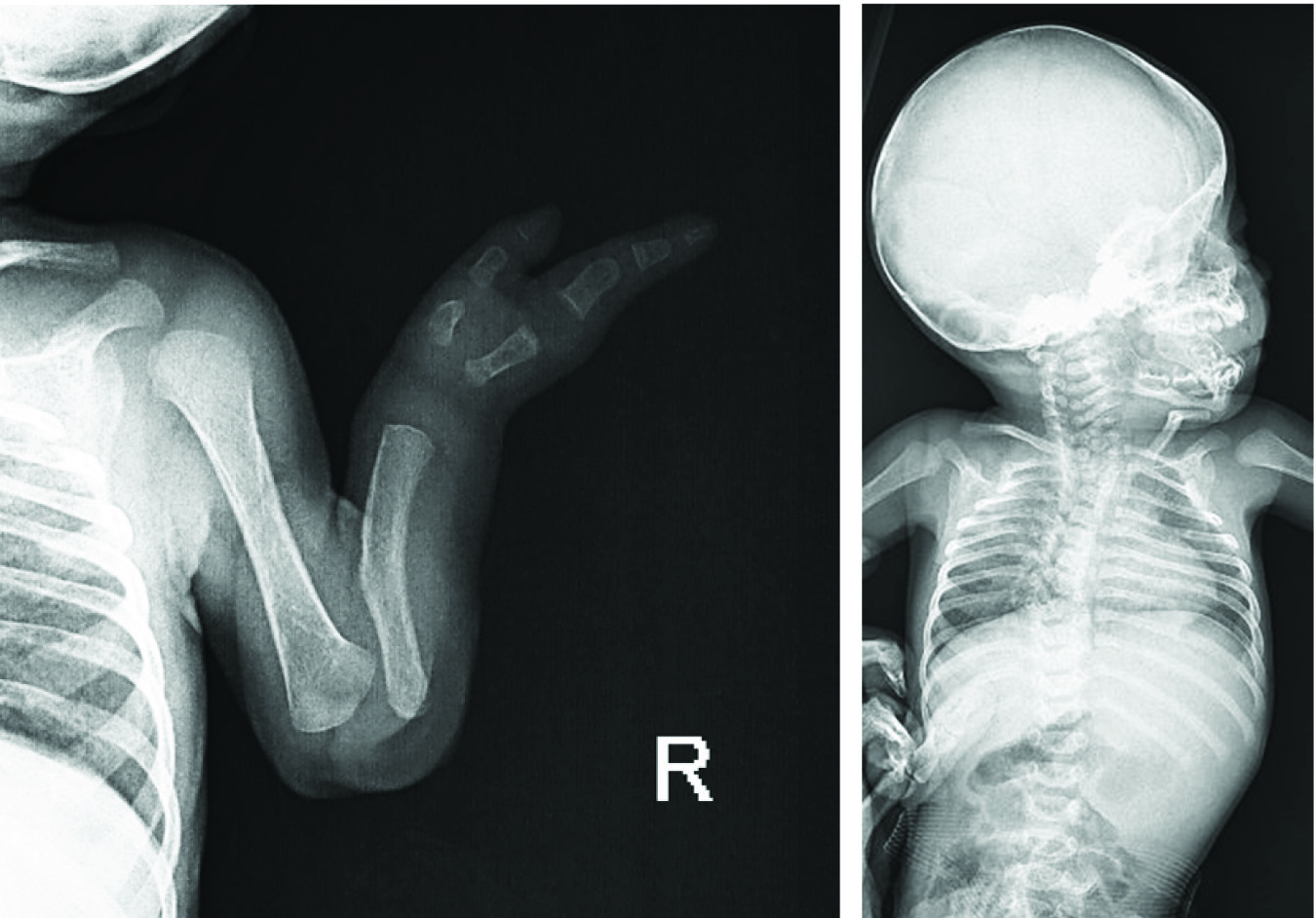
Bone survey showed the absence of radius and olygodactily of the right hand. A scoliosis with convexity to the right was also detected [Table/Fig-4]. Cranial magnetic resonance imaging was normal.
The graph depicting a. Absent Radius, olygodactily (absent digits) in her right hand b. Right-deviated scoliosis.
Discussion
Cornelia de Lange syndrome (CdLS), a multiple congenital anomaly syndrome, also called Brachmann-de Lange syndrome [1], is characterized by a distinguishing facial appearance, insufficient prenatal and postnatal growth, psychomotor delay, cognitive retardation, behavioral problems; abnormal upper extremities are also remarkable. Cardiac defects and gastrointestinal disorders are common, and many additional physical features occur, including myopia, palatal abnormalities, genitourinary abnormalities, pyloric stenosis, congenital diaphragmatic hernias and hearing loss. Facial dysmorphism includes arched eyebrows, synophrys, hirsute, short nose with anteverted nares, long philtrum, thin upper lip, and micrognathia which are among the main diagnostic features of CdLS. Autism and self-injurious tendencies also frequently occur. The exact incidence is unknown; it is estimated CdLS account for 1 in 10,000 newborns [2,3]. Although the genetic background is not clear, it is thought the main reason is the consequence of a dominant mutation in the NIPBL, SMC1L1 and SMC3 genes to date. However, a previous study in Iran highlighted the disease-causing role of the NIPBL mutations. Some CdLS cases seemed to be sporadic and 10% of the cases introduced chromosomal alterations, translocation of the 3q 26:2-q23, 5p13.2 [3–6].
Affecting many parts of the body, CdLS is a rare but a well characterized developmental disorder with varied features ranging from relatively mild to severe ones [7]. The disease severity is assessed based on a scoring system in which a score of more than 15 indicates moderate to severe disease [7,8].
According to the CdLS classification system suggested by Van allen et al., type I or classic CdLS patients display facial and skeletal alterations in the Preus & Rex diagnostic criteria [8]. They have prenatal growth deficiency; moderate to severe psychomotor developmental delay and major malformations, leading to vigorous disability or death. Type II or mild CdLS cases unveil similar facial and minor skeletal abnormalities than type I; however, these changes may develop over time or be partially manifested. They have mild to borderline psychomotor retardation, less severe pre/postnatal growth deficiency and the absence of (or less severe) major abnormalities. Type III or phenocopy CdLSs includes patients with CdLS phenotypic manifestations with causal relation to chromosomal aneuploidies or teratogenic exposures [9].
Generally, CDLS is featured by mental retardation related to a distinct series of physical malformations. Despite many physical manifestations occurring in a normal population [10], most cases described include severe structural deformations.
To date, some dental abnormalities have been reported such as eruption, spacing and macro- or microdontia. Yamamoto et al., have reported two cases with delayed tooth eruption and microdontia with one case of partial anadontia. Moreover, cardiovascular, endocrine and gastrointestinal abnormalities are considered possible [9].
The patient’s legal caretakers were included in this study after informed consent was obtained; based on the distinct facial features, physical findings and a normal karyotype, our patient was diagnosed as type I CdLS.
Excluding western countries, rare cases of CdLS have been reported worldwide.
Ellaithi et al., reported a case of CdLS from Sudan for the first time. Their patient was a seven-month-old girl who was diagnosed as the case of CdLS after investigating the reason of her refer which was malnutrition [1].
Tayebi introduced a case of CdLS from Iran for the first time. A 15-week-old and two-month-old boy who was referred as a multiple congenital anomalies that his clinical assessmentled to CdLS diagnosis [11].
Galehdari et al., has also reported a two-month-old boy with typical clinics of CdLS in Iran [6].
Despite previous cases reported in other regions of Iran, our patient is the first case of CdLS from Golestan province, northern Iran. Detailed features of the patients reported in the literature has been summarized and compared with other introduced cases in [Table/Fig-5].
Comparison of clinical features of CdLS in the present case clinical features frequency in CdLS Present case [3].
| Prenatal onset growth retardation (68%) + |
| Initial hypertonicity (100%) + |
| Low-pitched weak cry in infancy (74%) + |
| Feeding difficulties in the newborn period and infancy (71%) + |
| Microbrachycephaly (93%) + |
| Bushy eyebrows and synophrys (98%) + |
| Long, curly eyelashes (99%) + |
| Depressed nasal bridge (83%) + |
| Anteverted nares (85%) - |
| Down-turned angles of the mouth (94%) + |
| High arched palate (86%) |
| Micrognathia (84%) - |
| Spurs in the anterior angle of the mandible, prominent symphysis (66%) - |
| Short neck (66%) + |
| Hirsutism (78%) + |
| Low anterior and posterior hairline (92%) + |
| Hypoplastic nipples and umbilicus (50%) - |
| Micromelia (93%) - |
| Phocomelia and oligodactyly (27%) + |
| Clinodactyly of fifth fingers (74%) - |
| Simian crease (51%) + |
| Proximal implantation of thumbs (72%) - |
| Hypoplastic external genitalia (57%) + |
| Ophthalmologic manifestations (50%) - |
| Cutis marmorata and perioral pale cyanosis (56%) - |
| Seizures (23%) + |
| Congenital Heart Defect (33%) + |
“(%)” prevalence among the diagnosed cases “-/+ “presence in the current case.
Conclusion
Introducing CdLS cases of different ethnic backgrounds could add distinctions to the phenotypic picture of the syndrome and be useful in diagnosis. So then a proper diagnosis could lead to a better care and treatment. Our case has been under closer medical control since her diagnosis with monthly general podiatrists and specialists’ visit for better clinical advice to have the most comfortable life. In the mean time we introduce the report, she is in favorable and stable general condition of health and experiencing the best possible quality of life. Early diagnosis and decreased mortality rates are achievable through enhanced awareness and better care on this syndrome.
“(%)” prevalence among the diagnosed cases “-/+ “presence in the current case.
[1]. Ellaithi M, Gisselsson D, Nilsson T, Elagib A, Fadl-Elmula I, Abdelgadir M, A case of Cornelia de Lange syndrome from SudanBMC Pediatr 2007 7:6 [Google Scholar]
[2]. Preus M, Rex A, Definition and diagnosis of the Brachmann–de Lange syndromeAmerican journal of medical genetics 1983 16(3):301-12. [Google Scholar]
[3]. Uzun H, Senses DA, Uluba M, Kocabay K, A newborn with Cornelia de Lange syndrome: a case reportCases J 2008 1(1):329 [Google Scholar]
[4]. Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-BNature genetics 2004 36(6):631-35. [Google Scholar]
[5]. Strachan T, Cornelia de Lange Syndrome and the link between chromosomal function, DNA repair and developmental gene regulationCurrent opinion in genetics & development 2005 15(3):258-64. [Google Scholar]
[6]. Galehdari H, Monajemzadeh R, Nazem H, Mohamadian G, Pedram M, Identification of a novel de novo mutation in the NIPBL gene in an Iranian patient with Cornelia de Lange syndrome: A case reportJournal of Medical Case Reports 2011 5(1):242 [Google Scholar]
[7]. Nakanishi M, Deardorff MA, Clark D, Levy SE, Krantz I, Pipan M, Investigation of autistic features among individuals with mild to moderate Cornelia de Lange syndromeAm J Med Genet A 2012 158A(8):1841-47. [Google Scholar]
[8]. Van Allen MI, Filippi G, Siegel-Bartelt J, Yong SL, McGillivray B, Zuker RM, Smith CR, Magee JF, Ritchie S, Toi A, Clinical variability within Brachmann-de Lange syndrome: a proposed classification systemAm J Med Genet 1993 47(7):947-58. [Google Scholar]
[9]. Yamamoto K, Horiuchi K, Uemura K, Shohara E, Okada Y, Sugimura M, Cornelia de Lange syndrome with cleft palateInt J Oral Maxillofac Surg 1987 16(4):484-91. [Google Scholar]
[10]. Filippi G, The de Lange syndrome. Report of 15 casesClin Genet 1989 35(5):343-63. [Google Scholar]
[11]. Tayebi N, Cornelia de lange syndromeIndian Journal of Human Genetics 2008 14(1):23-26. [Google Scholar]