A Rare Case of Sporadic Inclusion Body Myositis (s-IBM)
Sourya Acharya1, Samarth Shukla2, Pritum Kitey3, Shameem Khan4, SN Mahajan5
1 Professor, Department of Medicine, JN Medical College DMIMS Univ.Sawangi (Meghe) Wardha, Maharashtra, India.
2 Professor, Department of Pathology, JN Medical College DMIMS Univ.Sawangi (Meghe) Wardha, Maharashtra, India.
3 Resident, Department of Medicine, JN Medical College DMIMS Univ.Sawangi (Meghe) Wardha, Maharashtra, India.
4 Resident, Department of Medicine, JN Medical College DMIMS Univ.Sawangi (Meghe) Wardha, Maharashtra, India.
5 Professor, Department of Medicine, JN Medical College DMIMS Univ.Sawangi (Meghe) Wardha, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sourya Acharya, Professor, Department of Medicine, JN Medical College DMIMS Univ. Sawangi (Meghe) Wardha- 442004, Maharashtra, India.
E-mail: souryaacharya@yahoo.co.in
Sporadic inclusion body myositis (s-IBM) is an acquired inflammatory myopathy. Clinical presentation is variable. The usual presentation is progressive weakness and atrophy of the arms and leg muscles, especially of the quadriceps femoris which is invariably affected. It is classified under inflammatory myopathies, along with polymyositis and dermatomyositis. We present a case of s-IBM who presented with gradually progressive quadriparesis with characteristic quadriceps wasting and weakness in neck flexors. Electromyography revealed myopathic potential and muscle biopsy revealed features of inclusion body myositis.
Atrophy, Dermatomyositis, Polymyositis, Quadriparesis
Case Report
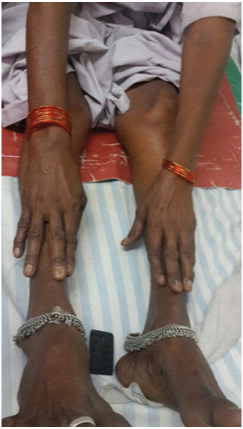
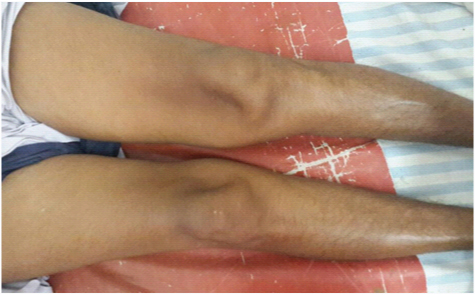
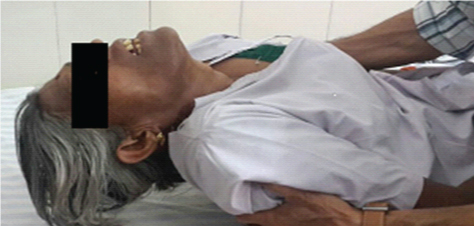
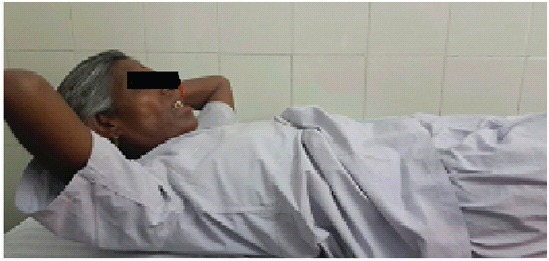
A 62-year-old female presented to medicine outpatient department with complains of gradually progressive quadriparesis of 7 years duration for which she had been investigated twice in some hospital, but no documents were available. The weakness started asymmetrically in the right leg and extended to involve all four limbs. Family history was negative for any similar illness. Her general physical examination was normal. Central nervous system examination revealed, normal higher functions, cranial nerves were normal, motor system examination revealed bilateral forearm and hand muscle wasting, bilateral quadriceps muscle wasting, leg wasting and feet wasting [Table/Fig-1,2]. Tone was normal. Power was grade 2/5 in bilateral wrist flexors, 3/5 in wrist extensors. Bilateral grip was 40%, Ankle dorsiflexors 3/5. Deep tendon reflexes (DTR) were preserved. Weakness of neck flexors revealed “drooped neck sign” and on attempted elevation of head the patient used both her hands to lift the head [Table/Fig-3,4]. Sensory examination was normal.
Showing bilateral peripheral muscle wasting.
Showing bilateral quadriceps wasting.
Showing drooping of head.
Showing the patient using hands on attempted elevation of head.
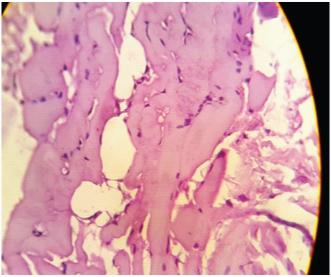
Investigations revealed normal complete blood counts, kidney and liver functions were normal, thyroid function was normal. Serum antinuclear antibodies, rheumatoid factor, vitamin D levels normal. Total CPK was elevated at 668 IU/L. EMG revealed low amplitude motor unit potentials with early recruitment suggestive of myopathy. A muscle biopsy from vastus lateralis showed features of inclusion body myosistis [Table/Fig-5]. The patient was empirically put on tablet prednisolone 40 mg once daily for one month and is awaiting follow- up.
H & E stained 40X slide showing vacuolations and a few vacuoles appear to be rimmed. Features suggestive of IBM.
Discussion
Sporadic inclusion body myositis (s-IBM) is an inflammatory myopathy which has got a wide variation in its clinical presentations, onset and prognosis [1,2].
The term was first coined in 1971 by Yunis and Samaha, when they detected vacuolations and inclusion bodies in muscle biopsy of a patient who presented as polymyositis [3]. The patients of IBM usually presents with diffuse, progressive, asymmetric, proximal, and distal weakness that is generally refractory to immunosuppressive treatment. Mean age of onset is 56-60 years [4,5].
Though the weakness variably affects the muscles, eventually both the proximal and distal muscles are involved. But involvement of quadriceps femoris and ankle dorsiflexors are characteristic of s-IBM. Dysphagia and mild facial weakness may occur but our patient did not have these symptoms. Isolated erector spinae weakness or "droopy neck" syndrome has been reported with s-IBM as in our case [6]. In most cases of s-IBM, serum CK level is normal or moderately elevated.
EMG reveals typically normal to reduced amplitude—reduced duration for simple (nonpolyphasic) motor unit action potentials (MUAPs). The presence of rimmed vacuoles is a characteristic feature of s-IBM [7]. The vacuoles occur singly or in multiples and are either subsarcolemmal or centrally located.
Inclusion body myositis usually does not respond to corticosteroids or other immunosuppressive drugs. Few patients improve slightly or cease worsening with steroids [8]. Another study suggested that low-dose methotrexate and corticosteroids may slow the progression of disease, but this regimen has not been widely adopted [9]. The disease in most patients is relentlessly progressive over many years, sometimes very slowly, and no method of treatment has so far altered the long-term prognosis.
Conclusion
Inclusion body myositis is the most common inflammatory myopathy seen in elderly. Usually it presents predominantly as an asymmetric gradually progressive quadriparesis with diminished deep tendon reflexes and dysphagia . Electromyography shows mixed myopathic and neurogenic changes. Muscle biopsy shows characteristic rimmed vacuoles. The aetiology of IBM is largely unknown. Immunopathology proposes a possible T cell-mediated inflammatory myopathy or a myodegeneration. Sporadic IBM is multifactorial as, genetic, immunological and environmental factors play a role in its development. No definitive treatment exists till now.
[1]. Argov Z, Hereditary inclusion body myopathies. In: Engel AG, Franzini-Armstrong C, edsMyology 2004 3rd edNew YorkMcGraw Hill:1311-20. [Google Scholar]
[2]. Malicdan MC, Noguchi S, Nishino I, Recent advances in distal myopathy with rimmed vacuoles (DMRV) or hIBM: treatment perspectivesCurr Opin Neurol 2008 21(5):596-600. [Google Scholar]
[3]. Yunis EJ, Samaha FJ, Inclusion body myositisLab Invest 1971 25(3):240-48. [Google Scholar]
[4]. Badrising UA, Maat-Schieman M, van Duinen SG, Epidemiology of inclusion body myositis in the Netherlands: a nationwide studyNeurology 2000 55(9):1385-87. [Google Scholar]
[5]. Phillips BA, Zilko PJ, Mastaglia FL, Prevalence of sporadic inclusion body myositis in Western AustraliaMuscle Nerve 2000 23(6):970-72. [Google Scholar]
[6]. Hund E, Heckl R, Goebel HH, Meinck HM, Inclusion body myositis presenting with isolated erector spinae paresisNeurology 1995 45(5):993-94. [Google Scholar]
[7]. Brady S, Squier W, Sewry C, Hanna M, Hilton-Jones D, Holton JL, A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositisBMJ Open 2014 4(4):e004552 [Google Scholar]
[8]. Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ, Inclusion body myositis. Observations in 40 patientsBrain 1989 112(Pt 3):727-47. [Google Scholar]
[9]. Sayers ME, Chou SM, Calabrese LH, Inclusion body myositis: analysis of 32 casesJ Rheumatol 1992 19(9):1385-89. [Google Scholar]