Crouzon Syndrome: Report in a Family
Dhanya S. Kumar1, Devaki Murugesan2, Kandasamy Murugan3, Divya Subramanian4, S. Uma Maheshwari5
1 Senior Lecturer, Department of Pedodontics and Preventive Dentistry, Rajas Dental College, Kavalkinaru, Tamil Nadu, India.
2 Senior Lecturer, Department of Pedodontics and Preventive Dentistry, Rajas Dental College, Kavalkinaru, Tamil Nadu, India.
3 Senior Lecturer, Department of Oral Medicine and Radiology, Rajas Dental College, Kavalkinaru, Tamil Nadu, India.
4 Senior lecturer, Department of Periodontics, Rajas Dental College, Kavalkinaru, Tamil nadu, India.
5 Senior Lecturer, Department of Conservative Dentistry and Endodontics, Rajas Dental College, Kavalkinaru, Tamil Nadu, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Dhanya S. Kumar, Senior Lecturer, Department of Pedodontics and Preventive Dentistry, Rajas Dental College, Kavalkinaru, Tamil Nadu, India.
E-mail: dhanya.sanalkumar@gmail.com
Correction:
This article has been updated on 1 March 2016, to see correction please see link which is given below:
http://jcdr.net/article_fulltext.asp?issn=0973-709x&year=2016&volume=10&issue=3&page=ZZ01&issn=0973-709x&id=7487
Craniosynostosis, Fibroblast growth factor receptor 2 gene
Craniosynostosis are a heterogeneous group of syndromes characterized by a premature sutural fusion that occurs individually or relating to other anomalies. Crouzon syndrome is the most common craniofacial dysostosis occurring without syndactyly [1]. This condition was first reported by a French Neurosurgeon, Octave Crouzon in 1912 in a mother and daughter [1].
A 13-year-old male patient reported to Rajas Dental College, Kavalkinaru with a chief complaint of pain in the lower right and left back tooth region since several days. The pain was chronic, aggravates on having food and intermittent. Medical history was noncontributory. There was a history of delayed achievement of developmental milestones. On extra oral examination, the patient had a brachycephalic skull with bilateral ocular proptosis, exopthalmos, mid face deficiency, relative mandibular prognathism, hypertelorism and low set ears and frontal bossing [Table/Fig-1a], [Table/Fig-1b].
(a) Frontal extra oral view;(b) Lateral profile;(c)Intra oral frontal view;(d)Mandibular occlusal view;(e)Maxillary occlusal view (f)Composite restorations done.
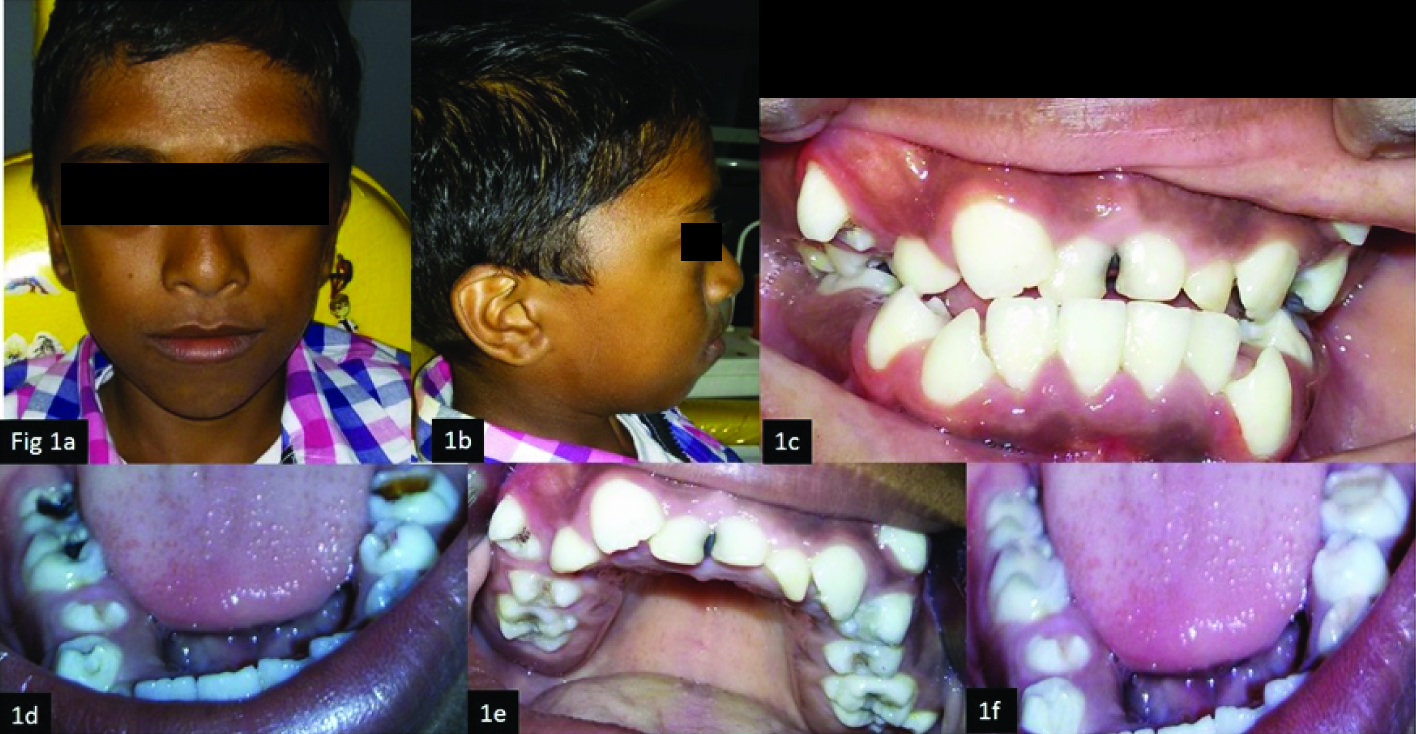
Lateral profile of the patient. No anomalies of upper or lower extremities were noted. On intraoral examination, retained 51, 61, 52 with clinically Missing 11,21; [Table/Fig-1c]. Deep caries in relation to 36, 46, 47 [Table/Fig-1d], moderate caries with 26, 16 [Table/Fig-1e].
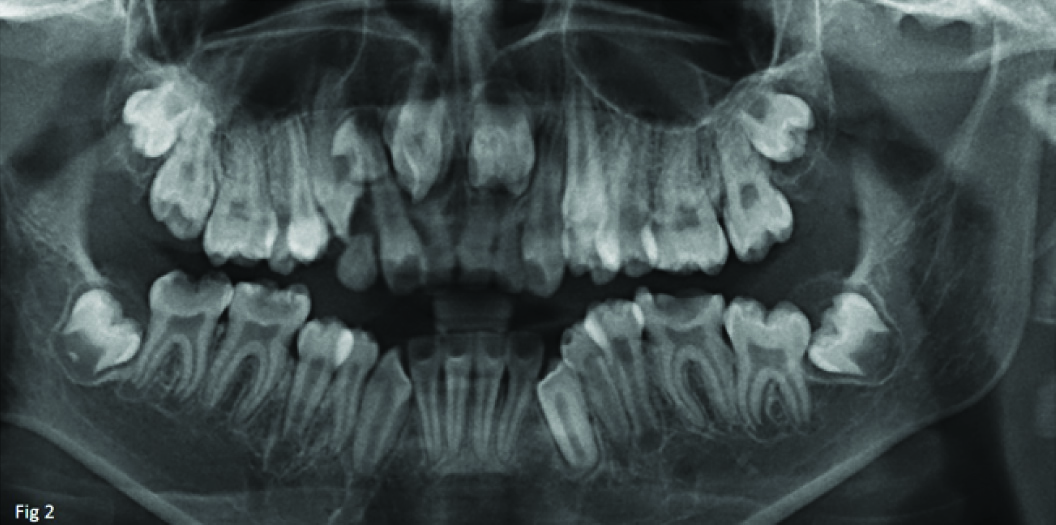
Deep pit and fissures with 44, 34, 24, 25, 14, 15. Edge to edge bite was evident. OPG revealed impacted 11, 21 [Table/Fig-2].
OPG of the patient showing impacted 11, 21.
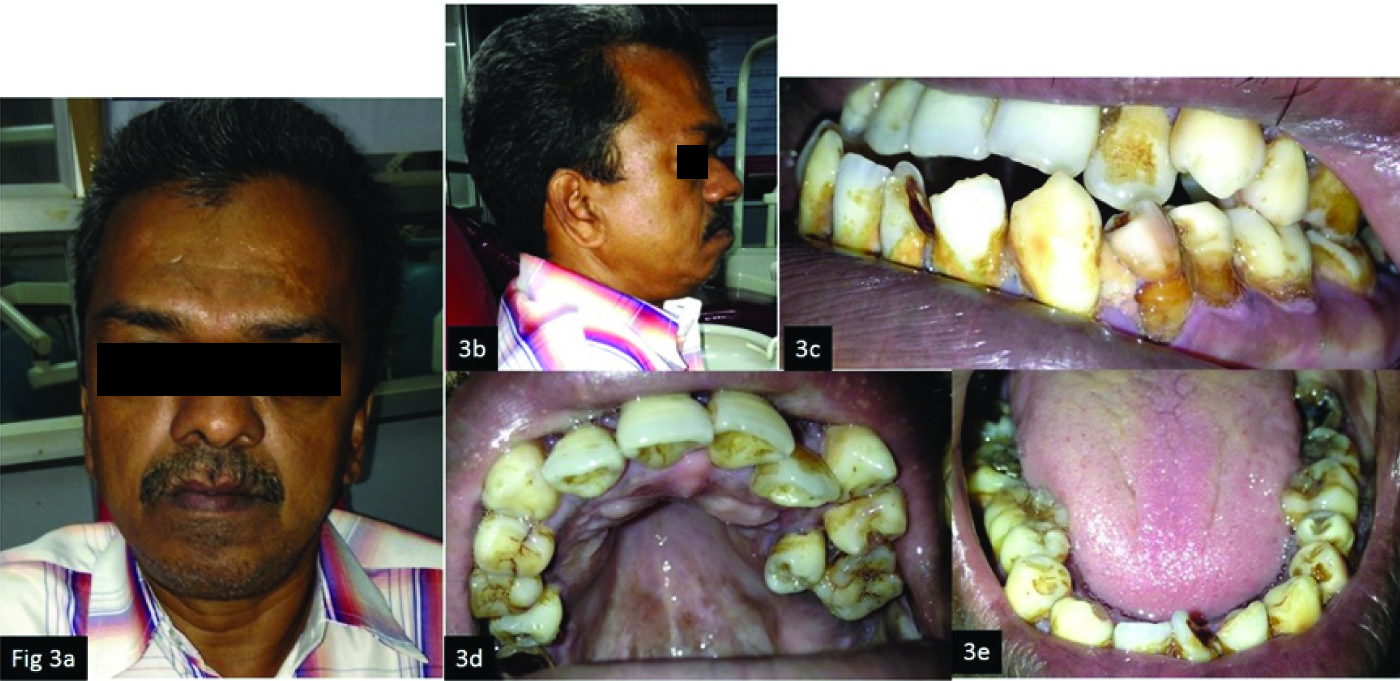
The patient’s father also had similar facial and intraoral features with anterior open bite and severe crowding [Table/Fig-3].
(a) Frontal extra oral view; (b) Lateral profile; (c) Intraoral view; (d) Maxillary occlusal view; (e) Mandibular occlusal view.
The clinical findings prompted a diagnosis of Crouzon syndrome. The patient was referred to pediatrician for detailed examination and confirmation of diagnosis.
Restoration of 16, 26, 36, 46, 47 was done with composite and pit and fissure sealants were applied on 44, 34, 24, 14, 15, 25; [Table/Fig-1f]. This patient also has retained 51, 61, 62 which requires extraction followed by orthodontic extrusion of impacted 11, 21, and combined orthodontic and surgical management for restoring the normal facial appearance.
Prevalence of this condition is 1 in 60000 live births. This occurs due to mutation in Fibroblast Growth Factor Receptor 2 (FGFR2) on chromosome locus 10q25-q26 resulting in early fusion of skull bones in fetal development [2–4]. The premature suture closure causes re-direction of skull growth parallel to the suture resulting in asymmetric shape of the skull. The characteristic cranial shapes are brachycephaly as seen in this patient, scaphocephaly, trigonocephaly and in severe disease, cloverleaf skull deformity [5].
Ocular proptosis, due to shallow orbits is a frequent finding which was evident in this patient as well. Other ocular anomalies include hypertelorism, strabismus, reduced monocular visual acuity and blindness [5]. Oral manifestations include an underdeveloped maxilla, which results in midface hypoplasia. This manifestation was noted to a mild degree in the present case. Ectopic eruption, crowding of maxillary teeth and mandibular teeth and anterior open bite are also commonly observed. Mental ability will be affected only if severe craniosynostosis occurs early in life and psychomotor development is generally normal [5].
Radiographic examination shows scelortic changes of sutures. Prominent cranial markings due to increased intracranial pressure may be seen as multiple radiolucencies, which results in a beaten metal appearance [5].
Differential diagnosis: Apert syndrome, Carpenters syndrome and Pfeiffers syndrome.
Management is multidisciplinary. Early diagnosis is very important as it is necessary to release the fused sutures to allow for normal brain development followed by skull reshaping for better esthetics. Also, combined orthodontic and surgical management will be required to provide normal appearance [6].
As a dentist we should know the clinical features which help in prompt diagnosis and better management of such children at a very young age. Patient’s written consent was obtained for the representation of the images.
[1]. Crouzon LE, Dysostose cranio faciale heriditaireBulletin de la societe des medicines deshospitaux de paris 1912 33:545-55. [Google Scholar]
[2]. Jeftha A, Stephen L, Morkel JA, Beighton P, Crouzonodermoskeletal syndromeThe journal of Clinical Pediatric Dentistry 2004 28:173-76. [Google Scholar]
[3]. Silva DL, Neto FX, Carneiro SG, Palheta AC, Monteiro M, Cunha SC, Crouzon’s Syndrome: Literature ReviewIntl Arch Otorhinolaryngol 2008 12:436-41. [Google Scholar]
[4]. Neville BW, Damm DD, Allen CM, Bouquot JE, Developmental defects of the oral and maxillofacial regionIn: Oral and maxillofacial pathology 2009 third editionElsevier:1-53. [Google Scholar]
[5]. Maloth S, Padamashree S, Rema J, Yalsangi S, Ramadoss T, Kalladka M, Diagnosis of Crouzon’s syndromeHong Kong Dent J 2010 7:95-100. [Google Scholar]
[6]. Kreiborg C, Birth prevalence studies of Crouzon syndromeComparison of direct and indirect methods Clin Genet 1992 41:12-15. [Google Scholar]