Accelerated Phase of Chediak-Higashi Syndrome at Initial Presentation: A Case Report of an Uncommon Occurrence in a Rare Disorder
Pooja Jaiswal1, Yogesh Kumar Yadav2, Nilam Bhasker3, Rashmi Kushwaha4
1 Assistant Professor, Department of Pathology, Integral Institute of Medical Sciences and Research, Lucknow, U.P., India.
2 Assistant Professor, Department of Pathology, Integral Institute of Medical Sciences and Research, Lucknow, U.P., India.
3 Resident, Department of Pathology, Integral Institute of Medical Sciences and Research, Lucknow, U.P., India.
4 Assistant Professor, Department of Pathology, King George Medical University, Lucknow, U.P., India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Yogesh Kumar Yadav, 145, A-Block, South City, Lucknow, UP, India.
E-mail: dr.yogi007@gmail.com
Chediak-Higashi syndrome (CHS) is an uncommon and fatal congenital disorder. The characteristic features of CHS are partial oculocutaneous albinism, increased vulnerability to infections, presence of abnormal large granules in leukocytes and an accelerated lymphohistiocytic phase. Accelerated phase at initial presentation is rarely seen as it is usually preceded by repeated episodes of infections. Hence this interesting case of a four-month-old Indian child born to consanguineous parents in accelerated phase at initial presentation is described. The boy presented with fever, hepatosplenomegaly, and cleft lip. Clinical diagnosis was leukemia or a lysosomal storage disorder. Cytopaenias, lymphohistiocytic infiltration in bone marrow, and the characteristic large granules in leucocytes helped in the diagnosis, emphasizing the importance of bone marrow in diagnosis of unusual presentation of this rare disorder.
Bone marrow aspiration, Congenital, Haemophagacytosis
Case Report
A four-month-old boy born of first degree consanguinous marriage was admitted in the paediatric ICU with complains of fever since 15 days and decreased feeding for one day. There was no history of rash or abdominal pain. He weighed 4 kilogram, was pale, febrile with pulse rate 138 beats per minute, and respiratory rate 38/minute. Physical examination showed blonde hair, fairer skin and cleft lip [Table/Fig-1]. Eyes were light brown in colour with nystagmus. Examination of the abdomen revealed hepatomegaly with a nontender liver, smooth surface, sharp border and span of 6.0 cm, but no jaundice clinically or lymphadenopathy. Spleen was palpable 5 cm below, the left costal margin. Cardiovascular, respiratory and central nervous system were normal.
Gross photograph showing blonde hairs and hypopigmented skin

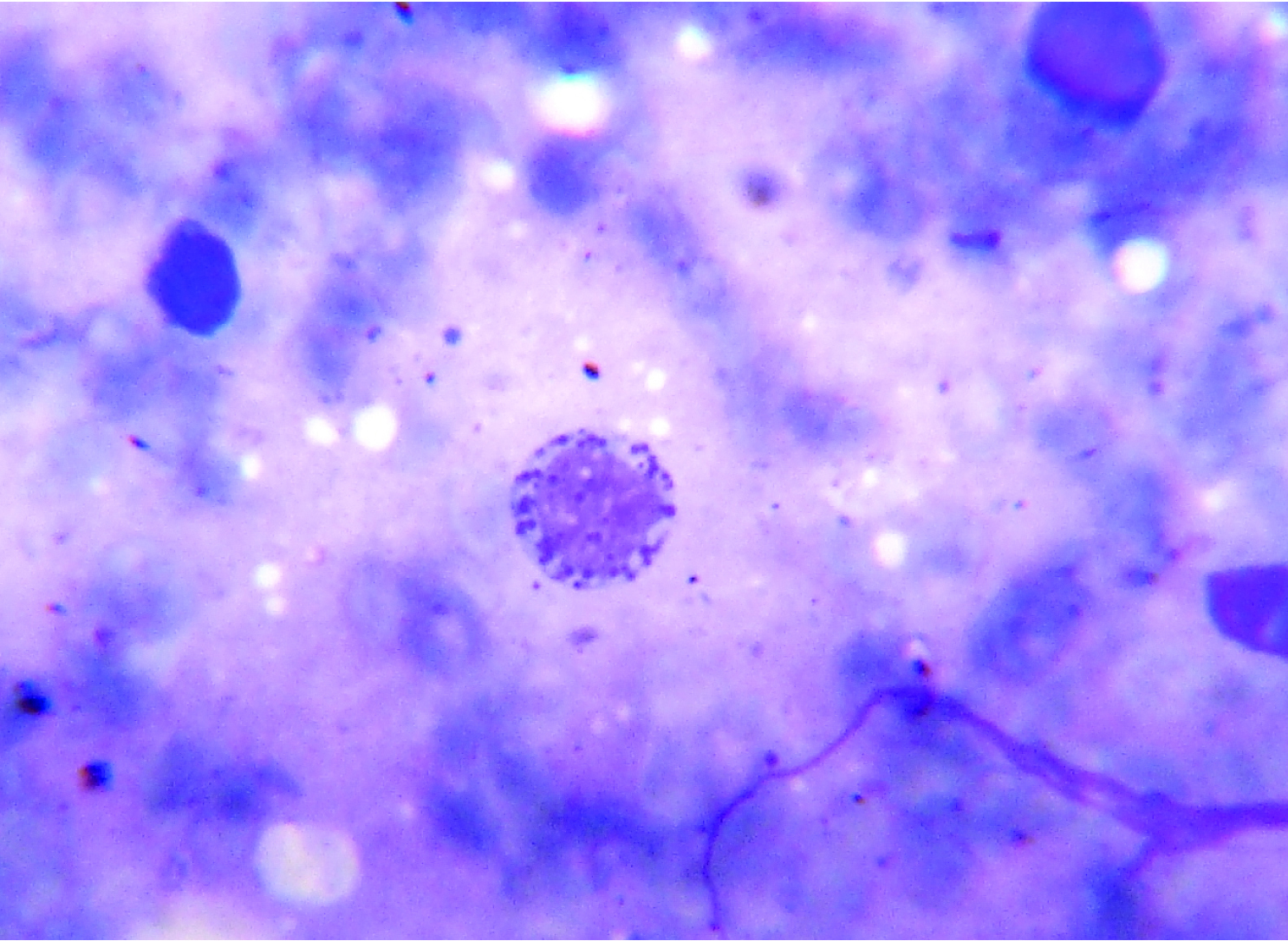
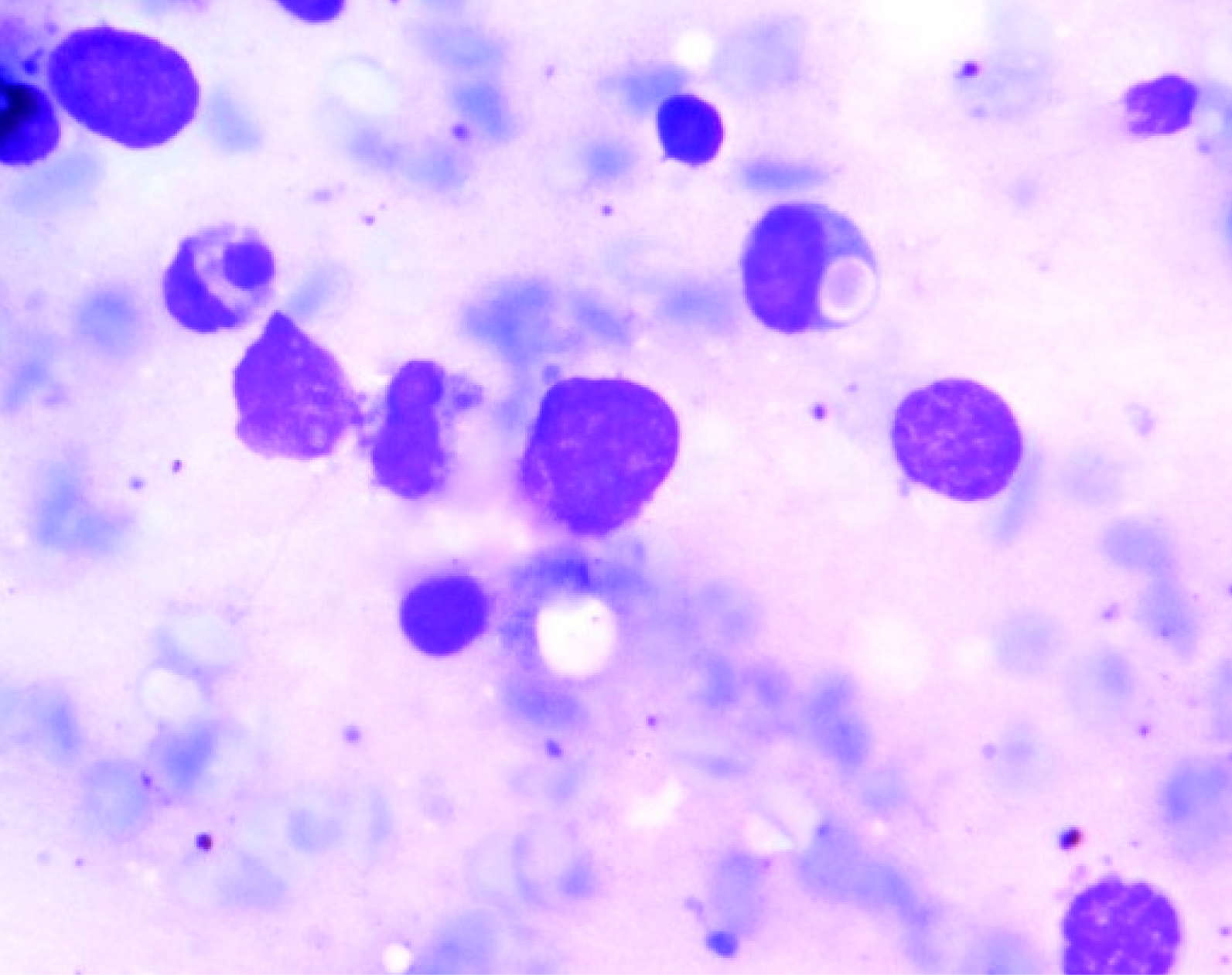
The haemogram findings were as follows: Hb 4.7 g/dl, a total leukocyte count of 7500 cells/μL, P05%, L94% and M01%; and ESR 9 mm at end of hour. Platelet count was 10,000/μL and reticulocyte count 0.5%. Blood group was O negative. The peripheral smear showed macrocytic red blood cells with moderate anisocytosis and fair number of smudge cells. The bone marrow smear showed hypercellular marrow, myeloid to erythroid ratio of 1:1, erythroid hyperplasia with megaloblastic change. Large granules in the cells of myeloid series [Table/Fig-2] and eosinophilic cytoplasmic inclusions were seen in myeloblast. Slightly increased histiocytes and haemophagocytic activity was noted [Table/Fig-3]. Megakaryocytes were decreased in number. No abnormal blast was seen in the smears examined. Liver Function Test and chest X-ray were within normal limits. Ultrasound abdomen showed hepatosplenomegaly. On ophthalmic examination normal fundus and retinal pigment deficit was noted. Serum ferritin was 530 ng/ml, cholesterol 193 mg/dl and triglycerides 187 mg/dl. Based on these findings, a diagnosis of accelerated phase of CHS was made.
Bone-marrow aspirate showing abnormal large intracytoplasmic granules in the myeloid precursors (Leishman Stain X 1000)
Bone-marrow aspirate showing haemophagacytosis (Leishman Stain X 1000)
The peripheral blood examination of the parents was unremarkable. The child was given symptomatic treatment in the form of antibiotics, platelet and packed cell transfusion for thrombocytopaenia and anaemia respectively. The child was referred to a higher centre for further management.
Discussion
Chediak-Higashi syndrome (CHS) is an uncommon and fatal autosomal recessive disorder affecting multiple system of body [1] characterized by partial oculocutaneous albinism, severe immune deficiency resulting in increased vulnerability to pyogenic infections, presence of abnormal large granules in leukocytes and an accelerated lymphohistiocytic phase. Accelerated phase at presentation is rarely seen [2] as it is usually preceded by repeated episodes of bacterial infections.
CHS was first described by Beguez-Cesar in 1943 and later by Steinbrinck in 1948, a Cuban hematologist, Chediak in 1952, and a Japanese paediatrician, Higashi in 1954. The first case in India was reported in 1982 [3]. To date about 500 cases have been reported in the world literature [4].
The average age of manifestation is about 6 years. In present case the child was four-month-old. Most patients die before the age of 10 year. CHS affects all the races. But it may be underreported in persons of darker-skinned races [5] as in this case finding of partial albinism was missed by the paediatricians. Fifty percent cases are found among children of parents with consanguineous marriages [3] as in this case.
The characteristic feature of CHS is the presence of huge lysosomes and cytoplasmic inclusions within different body cells like the white blood cells [6]. This is due to defect in granule morphogenesis caused by mutations of the lysosomal trafficking gene 2, first recognized in 1996 as the CHS1/LYST gene and is located on bands 1q of chromosome 1 [7] resulting in abnormal function of the lysosomal trafficking regulator protein which affect the size and function of lysosomes.
Clinical signs associated with this syndrome are hypopigmentation of the skin, eyes and hair; recurrent infection, easy bruisability, nystagmus and hepatosplenomegaly. Cellular and humeral immune deficiencies are seen in CHS [3] resulting in progression of viral and bacterial infections. Present case didn’t have any history of recurrent pyogenic infections. But the accelerated phase was the primary presentation in our case. The present case had blonde hair, fair skin, retinal hypopigmentation, nystagmus and hepatosplenomegaly.
About 50% of the cases develop neural manifestations like peripheral neuropathy, long tract signs, coma, convulsions and mental impairement [7]. In the present case patient didn’t have any neurological problem.
More than 75% of patients with CHS develop an accelerated phase of the disease also termed as haemophagocytic lymphohistiocytosis, is a hyperinflammatory syndrome with high mortality rate, characterized by fever, jaundice, hepatosplenomegaly, lymphadenopathy, pancytopaenia, coagulopathy, neurological abnormalities and diffuse mononuclear cell infiltrates into the organs which was noticed in present case.
According to the guidelines of Histiocytic Society last revised in 2004 [8] diagnosis of accelerated phase is based on the presence of a genetic defect, like mutation in LYST gene by sequence analysis, or five of the following features. These are: 1) history of unexplained, persistant or recurrent fever; 2) splenomegaly; 3) cytopaenias of at least two cell lineages; 4) hypertryglyceridaemia and/ or hypofibrinogedaemia; 5) evidence of haemophagocytosis in bone marrow; 6) low or absent NK-cell activity; 7) elevated serum ferritin levels (greater than 500 micro gram per liter); and 8) elevated CD25 levels (greater than 2400 U per milliliter). In present case fever, splenomegaly, mild hyperlipidaemia, anaemia, thrombocytopaenia, haemophagocytosis was seen.
The differential diagnosis includes other genetic disorders presenting with oculocutaneous albinism like Griscelli syndrome and Hermansky-Pudlak syndrome, but no giant granules seen in both the disorders. Giant granules resembling those seen in CHS may be seen as acute and chronic myeloid leukemia, also referred as pseudo-CHS anomaly [9]. The other diseases in differential diagnosis are Prader Willi and Angelman associated with hypopigmentation but without ophthalmic albinism.
Treatment of accelerated-phase of CHS is difficult and prognosis is poor [10]. Parentral vitamin C and bone marrow transplant has little benefit during this phase. Etoposide, steroids and intrathecal methotrexate have been tried in the accelerated phase [9].
Since the disease is autosomal recessive in transmission, screening of the patient’s parents by examination of the blood smears is recommended. Prenatal diagnosis is possible by demonstrating presence of positive phosphatase acid in lysosomes via amniotic fluid cells, CVS, and fetal blood leukocytes [11,12].
Conclusion
CHS is an uncommon and fatal disorder with variable clinical features and laboratory findings. Bone marrow transplant is the only curative treatment if performed early. It has little benefit during accelerated phase of CHS. Therefore peripheral blood examination and bone marrow aspiration are very important for early diagnosis and further management.
[1]. Westbroek W, Adams D, Huizing M, Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotypeJ Invest Dermatol 2007 127(11):2674-77. [Google Scholar]
[2]. Brown CC, Gallin JI, Chaemotactic disordersHematol Oncol Clin North Am 1988 2:61-79. [Google Scholar]
[3]. Seth P, Bhargava M, Kalra V, Chediak- Higashi syndromeIndian Paediatr 1982 19:950-52. [Google Scholar]
[4]. Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi Ch, Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associationsInt J Trichology 2011 3(2):107-11. [Google Scholar]
[5]. Al-Khenaizan S, Hyperpigmentation in Chediak-Higashi syndromeJ Am Acad Dermatol 2003 49(5):244-46. [Google Scholar]
[6]. Imran T, Zafar L, Rehan M, Nasir A, Tariq PA, Batool I, Chediak-Higashi syndrome presenting in accelerated phaseJ Coll Physians Surg Pak 2012 22(8):539-41. [Google Scholar]
[7]. Islam AS, Islam MS, Hawsawi SM, Unrelated Donor Marrow Transplantation for a Case of Chiediak-Higashi Syndrome with Hereditary ElliptocytosisIbrahim Med Coll J 2008 2:28-31. [Google Scholar]
[8]. Henter JI, Home A, Arico M, Egeler RM, Filipovich AH, Imashuku S, HLH-2004: Diagnostic and therapeutic guidelines for haemophagocytic lymphohistiocytosisPaediatr Blood Cancer 2007 48:124-31. [Google Scholar]
[9]. Skubitz KM, Qualitative disorders of leucocytes. In Lee GR, Foerster J, Lukens J, Paraskevas F, Greer JP, Rodgers GM. (Eds)Wintrobe’s Clinical Hematology 2009 12th edPhiladelphiaWilliams & Wilkins:1548-64. [Google Scholar]
[10]. Maaloul I, Talmoudi J, Chabchoub I, Ayadi L, Kamoun TH, Boudawara T, Chediak-Higashi syndrome presenting in accelerated phase: A case report and literature reviewHematol Oncol Stem Cell Ther 2015 :piiS1658-3876(15)00078-3. doi: 10.1016/j. haemonc. 2015.07.002. [Epub ahead of print] [Google Scholar]
[11]. Diukman R, Tanigawa S, Cown MJ, Globus MS, Prenatal diagnosis of Chediak-Higashi SyndromePrenat Diagn 1992 12(11):877-85. [Google Scholar]
[12]. Shyur SD, Hill HR, Recent advances in the genetics of primary immunodeficiency syndromesJ Paediatr 1996 129(1):8-24. [Google Scholar]