Co-Incidence or Co-Existence? Acute Lymphoblastic Leukaemia in HbE-alpha Thalassaemia: A Case Report with Review of Literature
Rithika Rajendran1, Febe Renjitha Suman2, Aruna Rajendran3, Julius Xavier Scott4
1 Postgraduate Student, Department of Pathology, Sri Ramachandra Medical College & Research Institute, Porur, Chennai, India.
2 Professor, Department of Pathology, Sri Ramachandra Medical College & Research Institute, Porur, Chennai, India.
3 Assistant Professor, Department of Paediatric Hematology Oncology, Sri Ramachandra Medical College & Research Institute, Porur, Chennai, India.
4 Professor, Department of Paediatric Hematology Oncology, Sri Ramachandra Medical College & Research Institute, Porur, Chennai, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Febe Renjitha Suman, Professor, Department of Pathology, Sri Ramachandra Medical College & Research Institute, Porur, Chennai-600 116, South India.
E-mail: febemd@gmail.com
Haemoglobin E (HbE) is a Haemoglobin variant that commonly occurs in many places in Asia. As β thalassaemia and α thalassaemia also occur in the same regions, the co-inheritance of these conditions leads to various phenotypic forms. HbE α thalassaemia is less common and of a milder phenotype than HbE β thalassaemia. Though malignancies are one of the complications in thalassaemia, occurrence of leukaemia is a rare event. Here we present a case of a two-year-old male child co-presenting with pre B acute lymphoblastic leukaemia (ALL) with MLL rearrangement and HbE alpha thalassaemia. The child is on remission 12 months post-therapy with standard ALL high risk protocol with no minimal residual disease (MRD). Haematological and oncological conditions coexisting at presentation is a challenge to therapy. This case is described for its rarity. Informed consent has been obtained from the parents.
Haemoglobin variants, Paediatric leukaemia, Thalassaemic syndrome
Case Report
A two-year-old male child, hailing from West Bengal, India, presented at Division of Paediatric Haemato-oncology, Department of Paediatrics, with complaints of frequent respiratory illnesses and decreased activity for the last 6 months. There was history of loss of appetite and weight loss since 2 months. The child with age appropriate developmental milestones was borne of a fourth degree consanguineous marriage. As preliminary investigations showed HbE α thalassaemia with Hb A2 –11.2 %, Hb F – 4.1% he had been referred to Sri Ramachandra Medical College Hospital for further management. On examination, general condition was fair, anthropometric measurements were within 95th percentile (Height - 87 cm, weight - 14 kg, head circumference – 45 cm). He had mild hepatosplenomegaly and multiple lymphadenopathy. Laboratory evaluation showed anaemia (Haemoglobin - 7.4 g/dL, RBC count - 2.54 x106/mm3), mild lymphomonocytosis (total count-13,400/mm3; lymphocytes 61.9%, monocytes 32.6%) and normal platelet counts (1,40,000/mm3). Peripheral blood film however showed microcytic RBCs with atypical lymphocytes and few blasts. Coomb’s test was negative. Serology for HIV, HBsAg, EBV and TORCH infections was negative. Since the peripheral smear showed blasts, a bone marrow examination was performed. Bone marrow aspirate showed 60% lymphoblasts. Bone marrow biopsy [Table/Fig-1] confirmed the diagnosis of ALL with increase in reticulin fibers. By immunohistochemistry the blasts were positive for CD45, CD34 and negative for CD10, CD 20 and TdT. Flow cytometric analysis showed that the blast population was strongly positive for CD19, CD34, HLA DR, CD45, weakly positive for CD22 and C79a and negative for CD10, TdT and myeloid markers. Cytogenetic analysis revealed MLL rearrangement by FISH. Repeat Haemoglobin variant analysis by HPLC showed Hb A2 –7.1% and Hb F – 4.3% suggestive of HbE α thalassaemia. A diagnosis of pre B acute lymphoblastic leukaemia with MLL rearrangement and HbE α thalassaemia was made. The mother was screened and tested negative for Hb variant analysis. Mutation analysis for thalassaemia and karyotyping of the patient as well as paternal screening could not be done.
Lymphoblast infiltrate in the bone marrow (H&E x400)
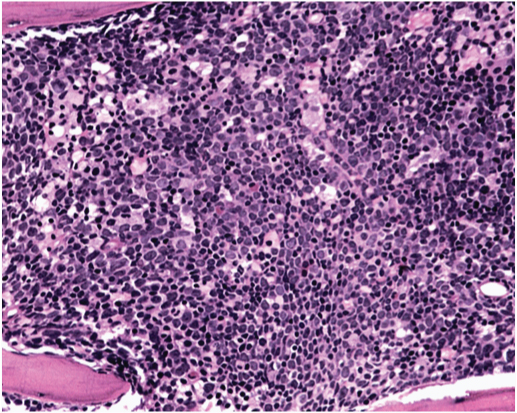
In view of the MLL mutation the child was categorized as high risk ALL. He received 4 cycles of induction, followed by intense consolidation and high dose methotrexate along with intrathecal methotrexate for CNS prophylaxis. Bone marrow was in morphological remission at the end of induction and was negative for MRD by flow cytometry. Cerebrospinal fluid was acellular. Induction phase was complicated by fungal pneumonia. The child completed delayed intensification phase of therapy and is on follow up. He is in remission since last 12 months.
Discussion
HbE disorders are one among the group of thalassaemic syndromes that occurs at higher frequencies in Asia and now increasing worldwide. Concomitant inheritance of α thalassaemia leads to HbE α thalassaemia, the clinical course of which varies according to the genotype. The prevalence of HbE/alpha thalassaemia in India is 0.06% [1]. Literature states that thalassaemic children develop complications including malignancies [2]. Malignancy is ranked fourth among the causes of death in thalassaemia [3].
Occurrence of leukaemia is rare with few cases of ALL and CML being reported in β thalassaemia [4–8]. However no case report of leukaemia associated with HbE thalassaemia or HbE α thalassaemia is available. All the three β thalassaemia major patients who developed pre B ALL are of paediatric age group and had multiple transfusions prior. The other β thalassaemia trait patient had T cell ALL who was cured of leukaemia [7]. Another case reported of a child with β thalassaemia intermedia and B cell ALL also achieved remission [8].
The present case is only 2 years old and had pre B ALL at time of diagnosis of HbE α Thalassaemia; the blasts are CD10 and TdT negative. TdT negative Pre B ALL is associated with CD10- CD34- blasts, high WBC count and MLL gene rearrangement [9]. It has also been reported that MLL rearrangement is associated with age <1 year, high WBC count and absence of CD 10 in leukemic blasts and CNS involvement [10]. But the present case had normal WBC count and the blasts were CD10- CD34+ with MLL rearrangement. The child had no CNS involvement.
Immune imbalance, generations of free radicals due to iron overload and recurrent infections in thalassaemic children have been considered as causes of leukaemia development [11,12]. However, the present case is very young and developed ALL at 2 years. Whether a genetic link may exist between the two diseases or is it only a co-existence need to be researched. The clinical features of leukaemia may be overlooked and repeated transfusions may mask anaemia when two Haematologic conditions co-exist. This may lead to a delay in the diagnosis of ALL.
Conclusion
Two haematological conditions may coexist; also pre B ALL can be TdT negative with CD34 positivity. MLL rearrangement is more common with TdT negative pre B ALL. The possibility of leukaemia is to be kept in mind when new symptoms like thrombocytopaenia develop in thalassaemic syndromes. Is this a pure coincidence or due to a combination of genetic, viral and environmental factors warrants further research in future.
[1]. Basu S, De R, HbE related Haemoglobinopathies, In. Agarwal MBHaematology today 2011 MumbaiAgarwal MB:147-160.Chapter 15 [Google Scholar]
[2]. Russo A, Schiliro G, Thalassaemia major and malignanciesAm J Haematol 1987 24:111-12. [Google Scholar]
[3]. Zurlo MG, De Stefano P, Borgna–Pignalti C, Di Palma A, Melevendi C, Piga A, Survival and causes of death in thalassaemia majorThe lancet 1989 334:27-40. [Google Scholar]
[4]. Lau B, Eife R, Lampert F, Acute lymphoblastic leukaemia after tuberculosis in a 8-year-old greek boy with homozygous beta –thalassaemiaKlinische Peadiatrie 1975 187:357-58. [Google Scholar]
[5]. Voskaridou E, Terpos E, Komninaka V, Chronic myeloid leukaemia with marked thrombocytosis in a patient with thalassaemia major: Complete haematological remission under the combination of hydroxyurea and anagrelideBr J Haematol 2002 116:155-57. [Google Scholar]
[6]. Nader M, Moghaddam EM, Alizadeh S, Acute lymphoblastic leukaemia in two patients with β thalassaemia majorZahedan J Res Med Sci (ZJRMS) 2014 10:29-30. [Google Scholar]
[7]. Derba S, Sathe AV, Pregnancy in survivor of acute lymphoblastic leukaemia with thalassaemia with Thalassaemia traitThe Journ Of Obstet. Gynae Of India 2012 62:S29-S30. [Google Scholar]
[8]. Tugcu D, Karakas Z, Gökçe M, Aoaoclu L, Ünüvar A, Sarıbeyoglu E, Thalassaemia Intermedia and Acute Lymphoblastic Leukaemia: Is it a Coincidental Double Diagnosis?Turk J Haematol 2014 31(3):311-12. [Google Scholar]
[9]. Liu L, McGavran L, Lovell MA, Wei Q, Jamieson BA, Williams SA, Non positive terminal deoxynucleotidyl transferase in paediatric precursor B-lymphoblastic leukaemiaArchives of Pathology Laboratory Medicine 2003 127:1340-44. [Google Scholar]
[10]. Behm FG, Raimondi SC, Frestedt JL, Liu Q, Crist WM, Downing JR, Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukaemia, regardless of presenting ageBlood 1996 87:2870-77. [Google Scholar]
[11]. Farmakis D, Giakoumis A, Polymeropoulos E, Aessopos A, Pathgenetic aspects of immune deficiency associated with beta thalassaemiaMed Sci Monit 2003 9:RA19-22. [Google Scholar]
[12]. Alavi S, Safari A, Sadeghi E, Amin S, Haematological malignancies complicating β thalassaemia syndromes: a single centre experienceBlood Res 2013 48:149-51. [Google Scholar]