Haemoglobin H disease, also known as the alpha-thalassaemia is characterized by the presence of HbH inclusions in red blood cells, detectable on supra-vital stain. We present a case of a previously asymptomatic 31-year-old male, who insidiously developed anaemia and had prominent splenomegaly. Peripheral smear examination revealed microcytic hypochromic anaemia with numerous spherocytes and moderate polychromasia. In reticulocyte preparation with Brilliant cresyl blue, HbH inclusions were mistakenly identified as granulofilamentous reticulum of reticulocytes, giving a spuriously high reticulocyte percentage. After the literature review, repeat assessment was performed and with the aid of high performance liquid chromatography result, it was possible to delineate the HbH inclusions.
Brilliant cresyl blue, Granulofilamentous reticulum, HbH inclusions, High performance liquid chromatography
Case Report
A 31-year-old male, previously asymptomatic, insidiously developed anaemia. He had prominent splenomegaly, 8 cm below the left costal margin and liver was palpable 2 cm below the right costal margin. A full blood count showed a haemoglobin concentration of 8.7 g/L, mean corpuscular volume (MCV) of 57 fL, mean corpuscular haemoglobin of 17pg, mean corpuscular haemoglobin concentration of 29% and red blood cell distribution width (RDW) of 28.3%; white blood cell count and differentials and platelet count were within normal limits (for age). Serum iron, ferritin and total iron binding capacity was within normal limits and lactate dehydrogenase was mildly elevated. Bilirubin levels mildly increased, with total and unconjugated being 1.9g/dL and 1.7g/dL, respectively. Peripheral blood smear examination revealed microcytic hypochromic red blood cell (RBC) morphology with tear drop poikilocytes, elliptocytes, target cells [Table/Fig-1a] and spherocytes [Table/Fig-1c,d: black arrow], along with 9 nucleated red blood cells in a background of moderate polychromasia [Table/Fig-1b:blue arrow]. On reticulocyte preparation with Brilliant cresyl blue, numerous inclusions [Table/Fig-1d:black arrow head] were noted in almost all RBCs. These inclusions and were mistakenly identified as granulo-filamentous reticulum [Table/Fig-1d:red arrow] of reticulocytes, resulting in an extremely high reticulocyte count of almost >95% [Table/Fig-1d].
Microcytic hypochromic RBC morphology with tear drop poikilocytes, elliptocytes, target cells [a] spherocytes [b: black arrow] and moderate polychromasia [b:blue arrow]. Reticulocyte preparation with Brilliant cresyl blue, numerous inclusions [d:black arrow head] were noted in almost all RBCs [c]. These inclusions and were mistakenly identified as granulo-filamentous reticulum of reticulocytes [d: red arrow]
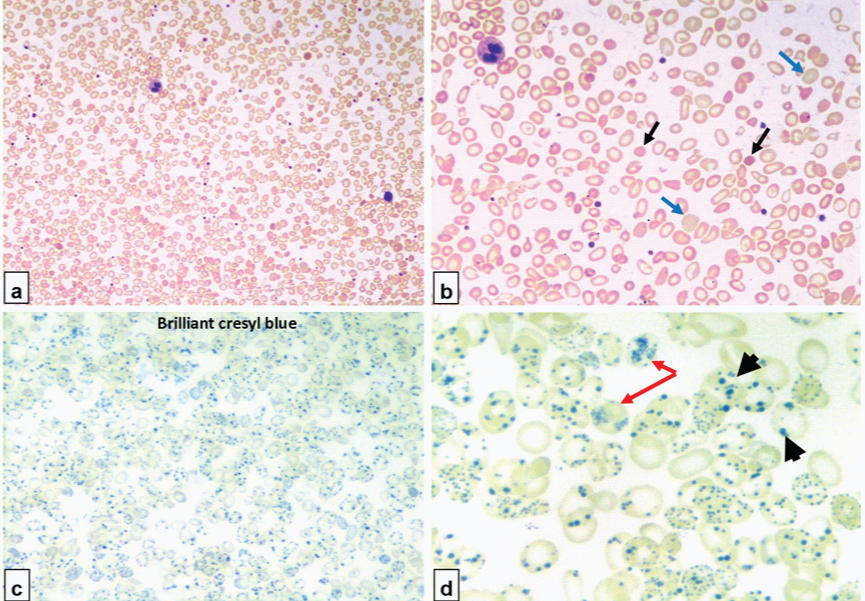
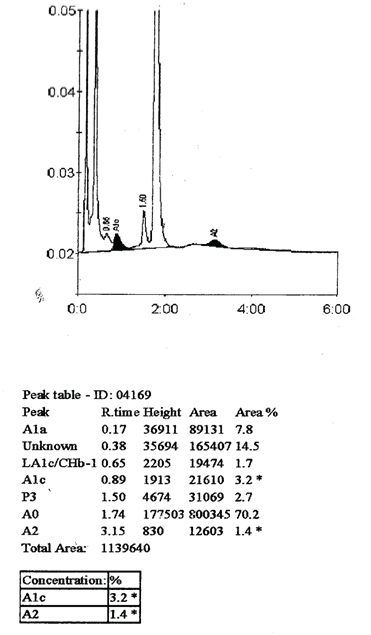
After review of clinical history and medical literature, the smear was scrutinized again pain stakingly to differentiate the reticulum from the inclusion. Reticulocyte count was finally established at 6.1%. Reticulum appears as granulo-filamentous and is deep blue in colour and HbH inclusion were small rounded bodies having a pale grey-blue colour. Sickling test was negative. High performance liquid chromatography to study types of haemoglobin was recommended, and it showed a significant decrease in HbA2 (1.4%; reference interval 2.5-3.4%) and HbA1c levels (3.2%; reference interval 4.2-6%). An unknown significant peak was observed at 0.38 minutes of retention time with an area of 14.5% [Table/Fig-2]. All these findings point towards presence of haemolysis and decreased HbA2 with significant peak in the first minute of elution along with the inclusion bodies on supravital stain point towards Haemoglobin H (HbH) disease. Parents of the patient had non-consanguineous marriage and his sibling brother had similar clinical features. His parents refused for their screening and further work-up. Developmental history was unremarkable. There was no prior history of any transfusion dependent anaemia. The patient was lost to follow-up.
High performance liquid chromatography revealed significant decrease in HbA2 (1.4 %; reference interval 2.5–34%) and HbA1c levels (3.2%; reference interval 4.2-6%). An unknown significant peak was observed at 0.38 minutes of retention timewith an area of 14.5%
Discussion
Haemoglobin H disease, an inherited haemoglobinopathy, is also known as the alpha (α)-thalassaemia [1]. Alpha-globin subunit is essential for both fetal and adult haemoglobin and responsible for oxygen transport in the blood [2]. Reduced α-globin production disrupts the ratio of globin chains in haemoglobin molecule. Deletions and non-deletional mutations affecting the adjacent α-globin genes, α1 and α2, on chromosome 16p13.3 result in reduced or absent-globin chain synthesis, causing α-thalassaemia [3]. In HbH disease, there is a relative excess of β chains forming a tetramer known as HbH (β4), and hence the name of the disease [3].
Haemoglobin H disease is generally a mild disorder. However, there is a marked phenotypic variability, ranging from asymptomatic presentations, to frequent need for periodic transfusions, to development of severe anaemia due to haemolysis with hepatosplenomegaly and cholelithiasis, and rarely even to fatal hydrops fetalis syndrome in utero [4]. While there is variability in the clinical course and presentations, some genotypes are sometimes associated with more severe disease course [4].
Haemoglobin H disease is the commonest form of thalassaemia intermedia and the most severe, non-fatal form of α-thalassaemia syndrome. It is caused due to decreased expression of α-globin [5]. In majority of cases, patients presents with compensated haemolytic anaemia, mild reduction in the haemoglobin that rarely requires blood transfusion, splenectomy or even standard iron chelation therapy. They are most often transfusion independent [5]. Hence, for long, this disease was clinically thought as a mild condition but in some patients there can be severe expression of HbH disease that may require regular transfusions and iron chelation therapy and therefore, HbH disease is not necessarily a benign disorder as previously thought [4,5].
Laboratory tests for the diagnosis of HbH disease include complete blood counts, peripheral smear examination, reticulocyte percentage, haemoglobin and DNA analysis. These patients have low MCV, MCH and a wide RDW. Red cell shape changes are similar to other thalassemic diseases characterized by moderate microcytic and hypochromic anaemia. Formation of HbH precipitates within red cells known as HbH inclusion bodies that can be induced by incubation with mild oxidants such as Brilliant Cresyl Blue. Numerous such inclusions in HbH disease and can easily be seen on light microscopy in most of the circulating erythrocytes. In our case, the HbH inclusion bodies were strikingly positive in all the erythrocytes. HbH can be detected by haemoglobin electrophoresis by various methods such as isoelectric focusing (IEF) or high performance liquid chromatography (HPLC). HbA2 level is usually down in the 1.0 -2.0% range [5]; our case the Hb A2 was reduced to 1.4%.
Non-deletional variants of HbH disease have more severe clinical course wherein growth retardation in childhood and iron overload in adults can be commonly encountered. The disease course will be severe in these patients and anaemia will be significant during infections, fever, hypersplenism, or pregnancy that ultimately may require blood transfusions [5].
HbH disease is a genetic disease where there is a 25% risk of acquiring the disease in each pregnancy when the partner carries a mutated α-globin gene [6]. Hence, genetic counseling and prenatal diagnosis is necessary with DNA-based genotyping. Not only the affected person and his partner, but also his other family members should undergo the screening of their carrier status. Southern blotting can be used for diagnosing deletional mutations. Gap-polymerase chain reaction (PCR) methods have been developed for many of the common deletions more recently [4]. Patients with HbH disease have risk of developing haemolytic crisis frequently during or after acute infections and high fever where the haemoglobin levels may drop to 3 g/dL overnight, because the red cells with precipitated HbH are rapidly destroyed. The patients can go into shock with acute renal failure. This is the only major complication in HbH disease that requires an immediate intervention with blood transfusions and treatment for infections [7].
A plastic crisis due to infection of Parvovirus B19 may occur in cases of HbH disease, but rarely reported. This condition should be considered in HbH patients who develop acute anaemia with reticulocytopenia and decreased white blood cells and platelet counts. Even though, Parvovirus B19 infection is self-limited disease that can undergo spontaneous resolution, however, at times it can be a critical emergency that requires a blood transfusion support. If the disease persists, bone marrow suppression is likely and in such cases, intravenous immunoglobulin should be considered [7].
Patients of alpha thalassaemia with milder form of disease do not require any specific therapy [8]. Splenectomy is always followed by significant elevation of haemoglobin levels in HbH disease, but it is not generally recommended because most patients do well with a steady state of haemoglobin levels. Splenectomy might be considered in patients with non-deletional HbH disease who have a more severe clinical phenotype and a baseline Hb of less than 7 g/dL, significant hepatosplenomegaly with or without hypersplenism, abdominal discomfort, and growth retardation that require frequent transfusion (10-12 transfusions per year) [9].
The pivotal role of supravital stain in the diagnosis of HbH is described earlier also, especially in the centers where molecular analysis is not done routinely due to cost constraints [10]. HbH disease is the most severe form of α-thalassaemia that is compatible with postnatal life and has a wide heterogeneity that can be attributed to the underlying genotype [11]. This disease is prevalent more commonly in the malarial belt and mutations and gene deletions resulting in thalassaemia genotype is independently seen in different ethnic communities [4]. The prevalence of α-thalassaemia in the Indian subcontinent varies ranging from 1%-18%. [12] A study conducted by Nadkarni et al., showed the varied genotypic heterogeneity. The author reported 5 cases of α0 thalassaemia (– –SA, – –SEA, – –20.5, – –MED, and – –KOL), 3 cases of α+ thalassaemia (–α3.7, –α4.2, and –α3.5) and 5 cases of non-deletional mutations (poly A tail, IVS1 nt 1 G → A, Hb Constant Spring, HbKoya Dora, and HbSallanches) [13]. The literature review states that only 3 to 4 genotypes have been documented in India leading to HbH disease. Due to limited number of cases and studies, it is difficult to look into the phenotypic variations of these HbH genotypes [12].
Conclusion
The diagnosis of the first case of HbH disease in any family requires a high index of suspicion as affected person is usually asymptomatic and diagnosis can be picked up on routine investigations only when one is aware of this entity. The diagnosis in our case was missed since the peripheral smear examination revealed non-specific microcytic hypochromic anaemia. Even in reticulocyte preparation with Brilliant cresylblue, HbH inclusions were mistakenly identified as granulo-filamentous reticulum of reticulocytes, giving a spuriously high reticulocyte percentage. Only on repeat assessment, and a look into the high performance liquid chromatography result, the diagnosis of HbH disease was possible. Following which, screening the other family members of the affected person was advised which has led to a proper genetic counseling and a possible prenatal diagnosis when required. A larger study about the incidence, natural history and clinical manifestations of HbH disease can definitely aid for a better insight into the darker unseen aspects of the disease.
[1]. Chan V, Wong MS, Ooi C, Chen FE, Chim CS, Liang RH, Can defects intransferrin receptor 2 and hereditary haemochromatosis genes account for ironoverload in HbH disease?Blood Cells Molecules & Diseases 2003 30:107-11. [Google Scholar]
[2]. Forget BG, Molecular Genetics of the Human Globin GenesIn: Disorders ofHaemoglobin; Genetics, Pathophysiology, and Clinical Management. Steinberg MH, Forget BG, Higgs DR, Nagel RL (eds) 2001 Cambridge, United KingdomCambridge University Press:117-30. [Google Scholar]
[3]. Higgs DR, Bowden DK, Clinical and Laboratory Features of α-Thalassaemia SyndromesIn: Disorders of Haemoglobin; Genetics, Pathophysiology, and Clinical Management. Steinberg MH, Forget BG, Higgs DR, Nagel RL (eds) 2001 Cambridge, United KingdomCambridge University Press:431-69. [Google Scholar]
[4]. Chui DHK, Fucharoen S, Chan V, Haemoglobin H disease: not necessarily a benign disorderBlood 2003 101:791-800. [Google Scholar]
[5]. Fucharoen S, Viprakasit V, Hb H disease: clinical course and disease modifiersHematology Am Soc Hematol Educ Program 2009 2009:26-34. [Google Scholar]
[6]. Cooley’s anaemia foundation Web site. http://www.cooleysanaemia.org/updates/pdf/Alpha_Thalassaemia.pdf. Accessed July 30, 2015 [Google Scholar]
[7]. Jetsrisuparb A, Sanchaisuriya K, Fucharoen G, Development of severeanaemia during fever episodes in patients with haemoglobin E trait and haemoglobinH disease combinationsJ Pediatr Hematol Oncol 2006 28:249-53. [Google Scholar]
[8]. Alpha Thalassaemia treatment & Management - Medscape Web site. http://emedicine.medscape.com/article/955496-treatment. Updated July 13, 2015. Accessed July 30 2015 [Google Scholar]
[9]. Al-Salem AH, Nasserulla Z, Splenectomy for children with thalassaemiaInt Surg 2002 87:269-73. [Google Scholar]
[10]. Kulaç I, Ünal S, Gümrük F, Brilliant cresyl blue staining for screening haemoglobin H disease: reticulocyte smearTurk J Hematol 2009 26:45 [Google Scholar]
[11]. Weatherall DJ, Clegg JB, The α-ThalassaemiasIn: The Thalassaemia Syndromes 1981 OxfordBlackwell Scientific Publications:508-612. [Google Scholar]
[12]. Nadkarni AH, Nair SB, Italia KY, Molecular Diversity of Haemoglobin H Disease in IndiaAm J Clin Pathol 2010 133:491-94. [Google Scholar]
[13]. Nadkarni A, Phanasgaonkar S, Colah R, Prevalence and molecular characterization of α-thalassaemia syndromes among IndiansGenet Test 2008 12:177-80. [Google Scholar]