Inheritance of Hereditary Persistence of Fetal Haemoglobin (HPFH) in a Family of Western Odisha, India
Siris Patel1, Snehadhini Dehury2, Prasanta Purohit3, Satyabrata Meher4, Kishalaya Das5
1 Senior Medical Officer, Sickle Cell Clinic and Molecular Biology Laboratory, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Odisha, India.
2 Research Assistant, Sickle Cell Clinic and Molecular Biology Laboratory, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Odisha, India.
3 Senior Research Fellow, Sickle Cell Clinic and Molecular Biology Laboratory, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Odisha, India.
4 Research Assistant, Sickle Cell Clinic and Molecular Biology Laboratory, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Odisha, India.
5 Scientific Officer, Sickle Cell Clinic and Molecular Biology Laboratory, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Odisha, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Prasanta Purohit, Senior Research Fellow (ICMR), Sickle Cell Clinic and Molecular Biology Laboratory, Room No. 11, OPD Building Veer Surendra Sai Institute of Medical Sciences and Research, PO-Burla, Dist. Sambalpur, Odisha-768017, India.
E-mail: prasanta.biochem@gmail.com
Hereditary persistence of foetal haemoglobin (HPFH) is a rare inherited haemoglobin disorders in India. We encountered five cases of HPFH-3 in heterozygous condition in a single family of western Odisha, India. All the cases had raised % HbF (26.1±3.23%) with pancellular distribution of HbF in erythrocytes. There were no abnormalities found in the red cell indices. All the cases were asymptomatic till date with normal growth and development. Molecular confirmation of this haemoglobin disorders is important for control and prevention of haemoglobinopathies in this region.
Alpha thalassemia, Haemoglobin, HPFH
Case Report
A 18-year- Old female of caste “Chasa” with mild fever had attended to the Sickle Cell Clinic for screening of sickle cell status. All the haematological and molecular investigations were done at Sickle Cell Clinic and Molecular Biology Laboratory, Veer Surendra Sai Institute of Medical Sciences and Research, Burla, Odisha, India. On investigation, her haemoglobin (Hb) was 12.9 g/dL with other blood parameters being normal. Sickling slide test was negative with “AF” pattern in agarose gel Hb electrophoresis (pH 8.6). Complete blood count was analysed by Sysmex KX-21; Sysmex Corporation, Kobe, Japan. Reticulocytes count was estimated by flow cytometer (BD Facs Caliber) using Retic Count reagent/thiazole orange reagent by using FACS Comp software as per the guideline. Distribution of HbF in the red cells was analysed by Kleihauer–Betke’s slide test. Hb fractions was quantified by high performance liquid Chromatography (CE-HPLC) by using the VARIANT-II Hb testing system; (Bio-Rad Laboratories, Hercules, CA), resulting a high HbF level of 25.5%. Her family screening revealed a high HbF level in other four members in the family with a mean HbF level of 26.1±3.23%.
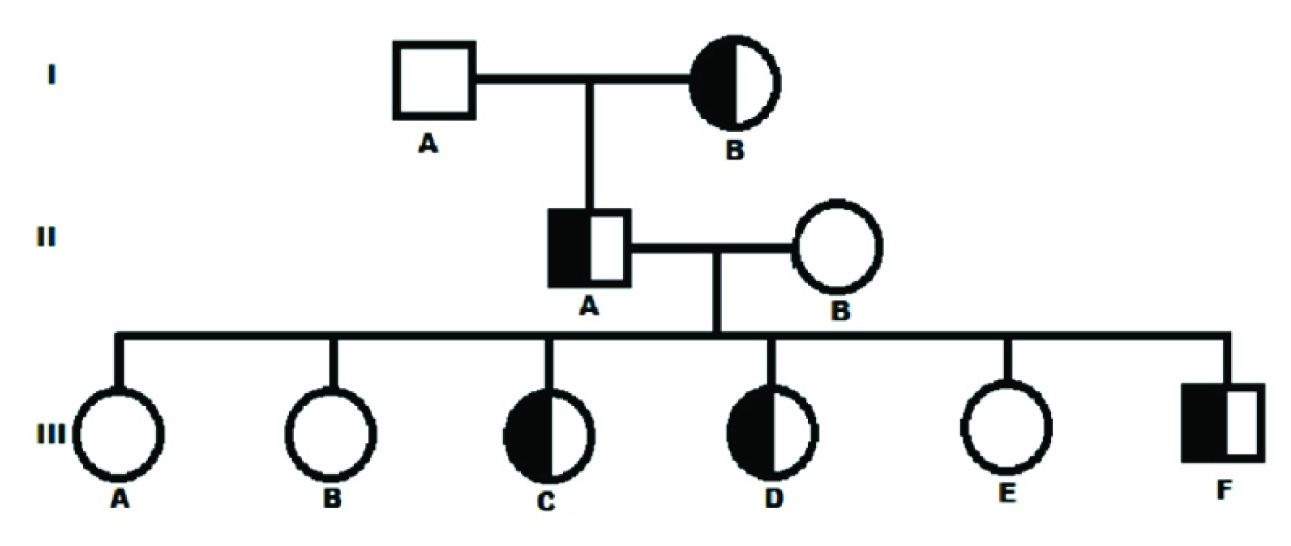
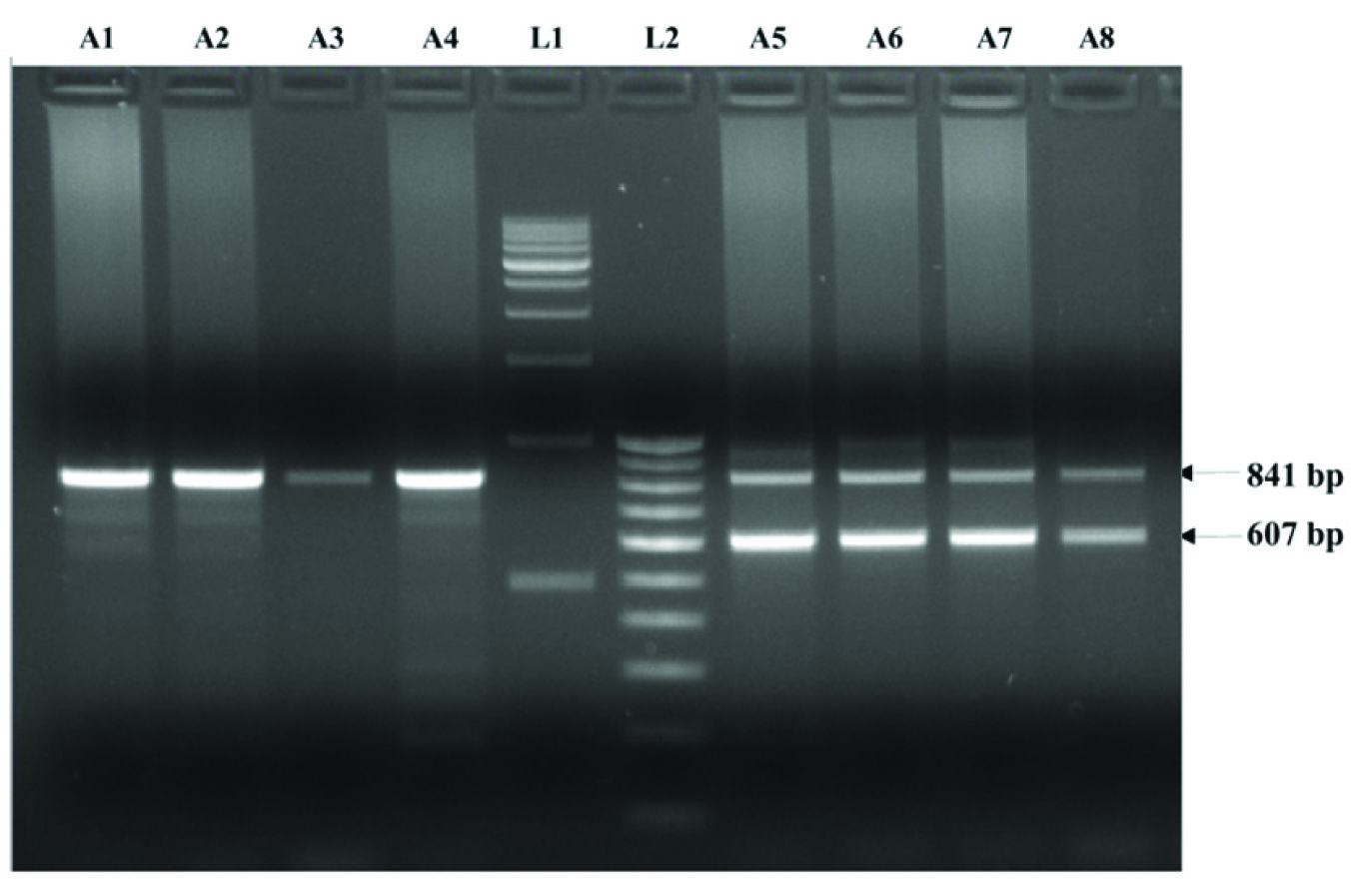
Complete pedigree was made carefully as per their genotypes [Table/Fig-1]. All the cases were asymptomatic till date and, had normal haematological reports. DNA isolation from peripheral blood was carried out by phenol-chloroform method. Molecular analysis for high HbF was done by procedures described earlier [1]. Both Asian Indian inversion-deletion GγAγ(δβ)0 and HPFH-3 (NG_000007,3:g,50509_83170del32662), the most prevalent high HbF determinants in India were investigated. All the five cases with high % HbF were found to be heterozygous HPFH-3. Agarose gel electrophoresis for molecular confirmation of HPFH-3 was shown in [Table/Fig-2]. Alpha thalassemia was investigated by GAP-PCR [2]. Only -3.7kb deletional alpha thalassemia was found in heterozygous condition in three members of the family (IIB, IIIB and IIIE). None of the case of heterozygous HPFH-3 had alpha thalassemia. Cases with alpha thalassemia had microcytic-hypochromic blood picture compared to others. All the HPFH-3 heterozygous cases had normal red cell indices. Pancellular distribution of HbF in red cells was found in all the HPFH-3 heterozygous cases. Reticulocytes were in normal range in all the cases. The haematological and molecular characterization of all the family members was described in [Table/Fig-3]. Written informed consent was obtained from the cases. This study was approved by Institutional ethical committee.
Pedigree showing inheritance of hereditary persistence of foetal hemoglobin in heterozygous condition
Agarose gel electrophoresis showing deletional high persistence of foetal hemoglobin (HPFH-3). Amplifaction of 841 bp PCR product is for normal and 607 bp for deletional HPFH-3. Lane A1, A2, A3 and A4 are normal and Lane A5 A6, A7 and A8 are heterozygous for HPFH-3, lane L1 and L2 are 500bp and 100 bp DNA ladder
Haematological and molecular characterization of HPFH-3 cases
| Parameters | IB | IIA | IIB | IIIA | IIIB | IIIC | IIID | IIIE | IIIF |
| Age (in years) | 73 | 50 | 43 | 26 | 24 | 22 | 18 | 15 | 12 |
| Relationship | GM | F | M | D1 | D2 | D3 | D4 | D5 | S1 |
| Haemoglobin fraction by HPLC |
| HbA0 (%) | 65.9 | 67.4 | 87.9 | 88.9 | 88.3 | 61.3 | 65 | 86.9 | 61.7 |
| HbA2 (%) | 2.0 | 2.3 | 3.1 | 2.7 | 2.8 | 2.1 | 2.2 | 2.7 | 2.3 |
| HbF (%) | 25.2 | 21.8 | 0.4 | 0.4 | 0.3 | 30.5 | 25.4 | 1.0 | 27.7 |
| Red cell indices |
| WBC (103/μL) | 7.6 | 5.6 | 4.2 | 6.5 | 8.1 | 7.9 | 7.8 | 7.1 | 6.8 |
| RBC (106/μL) | 4.69 | 6.10 | 3.33 | 4.19 | 3.33 | 5.55 | 4.92 | 5.72 | 5.64 |
| Haemoglobin (g/dL) | 9.8 | 15 | 11.8 | 12.5 | 13.7 | 14.1 | 12.9 | 12.2 | 13.3 |
| HCT (%) | 35.1 | 48.3 | 32 | 37.9 | 29 | 43.7 | 38.5 | 40.3 | 41.4 |
| Platelets (103/μL) | 172 | 266 | 236 | 249 | 222 | 278 | 168 | 283 | 183 |
| MCV (fL) | 79.8 | 79.2 | 70.2 | 90.5 | 72.6 | 78.7 | 78 | 70 | 76.4 |
| MCH (pg) | 26.2 | 24.6 | 22.2 | 30.1 | 23.6 | 25.4 | 26.1 | 21.3 | 23.6 |
| MCHC (g/dL) | 31.4 | 31.1 | 31.6 | 33.2 | 28.9 | 32.3 | 33.4 | 30.2 | 32.1 |
| Other Investigations |
| Reticulocyte count (%) | 0.34 | 0.86 | 0.74 | 0.54 | 0.48 | 0.84 | 0.72 | 1.03 | 0.76 |
| Distribution of HbF | Pan | Pan | - | - | - | Pan | Pan | - | Pan |
| Alpha thalassemia | αα/αα | αα/αα | -3.7α/αα | αα/αα | -3.7α/αα | αα/αα | αα/αα | -3.7α/αα | αα/αα |
| HPFH-3 | A/HPFH | A/HPFH | A/A | A/A | A/A | A/HPFH | A/HPFH | A/A | A/HPFH |
*WBC, white blood cells; RBC, Red blood cells; HCT, Haematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular Hb; MCHC, mean corpuscular Hb concentration; Pan–pancellular; GM, Grandmother; F, father; M, mother, D, daughter; S, Son. HPFH, High persistence of foetal haemoglobin
Discussion
Hereditary persistence of foetal haemoglobin (HPFH) are the condition caused by a large deletions involving both δ- and β-globin genes in the β-globin cluster, with increased level of HbF levels in adults [3]. In HPFH-3, the deletion removes 48.5 kb of a DNA segment, starting from the 5’ end of the ψβ gene to a region 30 kb downstream of the β-globin gene in an L1 repetitive region. This genetic trait, although rare, have been reported from different parts of India [4–6]. In literature, seven various forms of deletional HPFH have been reported. Out of which, HPFH-3 causing 48.5 kb deletion in the β globin gene cluster is the commonest form reported in India. The present patient-based diagnostic study reveals five cases of heterozygous HPFH-3 in a single family of western Odisha. Presence of high %HbF (26.1±3.23%; range 21.8 -30.5%) and normal red cell indices in all these five HPFH-3 heterozygous cases were similar to previous studies on HPFH reported in India [4–6]. The finding of a homogeneous HbF distribution in red cells was in agreement to the earlier report [6].
Deletional HPFH syndromes with a pancellular distribution of HbF have been reported to present benign in heterozygous state, with or without presence of other FC loci [7]. Our study agrees to this and all five cases were asymptomatic except the index (III D), who attended to the Sickle Cell Clinic and Molecular Biology Laboratory with mild fever. On the contrary as co-inheritance of HPFH with β thalassemia or HbE usually lead to severe clinical manifestation similar to that of thalassemia intermedia [8]. It becomes essential to study the allochtonous Hb disorders like the HPFH when introduced to an indigenous populations with established high frequency of HbS and β thalassemia alleles. This unique situation is particularly applicable to the western part of the province of Odisha situated in eastern India, with a reportedly high frequency of HbS and β thalassemia carrier (21% and 3.75 % respectively) and sporadic cases of HbE [9].
We have a limitation in this study that the information provided here caters to only the population of Odisha. A guideline for population based screening of such haemoglobin disorders should be necessary to generate data on national level will be a step forward for preventing and controlling program for haemoglobinopathies.
Conclusion
Molecular characterization of HPFH is important for understanding the resulting patho-physiology of the phenotype and they will also provide the information on the cis-acting genetic regulatory elements present in the β-globin cluster. Besides, high HbF determinants like HPFH are vital for prenatal diagnosis to prevent the affected foetus with obvious clinical manifestation when inherited with other forms of haemoglobin disorders. Indexing various forms of HPFH based on the molecular diagnosis in countries like India with a mosaic of endogamous communities and specific cultural practices will facilitate prevention and control program for haemoglobinopathies.
*WBC, white blood cells; RBC, Red blood cells; HCT, Haematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular Hb; MCHC, mean corpuscular Hb concentration; Pan–pancellular; GM, Grandmother; F, father; M, mother, D, daughter; S, Son. HPFH, High persistence of foetal haemoglobin
[1]. Craig JE, Barnetson RA, Prior J, Raven JL, Thein SL, Rapid detection of deletions causing δβ thalassemia and hereditary persistence of fetal haemoglobin by enzymatic amplificationBlood 1994 83(6):1673-82. [Google Scholar]
[2]. Chong SS, Boehm CD, Higgs DR, Cutting GR, Single-tube multiplex- PCR screen for common deletional determinants of alpha-thalassemiaBlood 2000 95:360-62. [Google Scholar]
[3]. Mayuranathan T, Rayabaram J, Das R, Arora N, Edison ES, Chandy M, Identification of rare and novel deletions that cause (δβ)0-thalassaemia and hereditary persistence of foetal haemoglobin in Indian populationEur J Haematol 2014 92(6):514-20. [Google Scholar]
[4]. Ghosh K, Hasan SW, Kumar S, Hereditary persistence of foetal haemoglobin in northwest IndiaTrop Geogr Med 1988 40(3):209-12. [Google Scholar]
[5]. Balgir RS, Hereditary persistence of foetal haemoglobin in a tribal family of Orissa, IndiaNatl Med J India 2004 17(3):138-40. [Google Scholar]
[6]. Nadkarni A, Wadia M, Gorakshakar A, Kiyama R, Colah RB, Mohanty D, Molecular characterization of δβ-thalassemia and hereditary persistence of fetal haemoglobin in the Indian populationHaemoglobin 2008 32(5):425-33. [Google Scholar]
[7]. Galanello R, Cao A, Relationship between genotype and phenotype. Thalassemia intermediaAnn N Y Acad Sci 1998 850:325-33. [Google Scholar]
[8]. Weatherall DJ, Clegg JB, The Thalassaemia Syndromes, chapter 10 2001 4th editionOxford, UKBlackwell Science [Google Scholar]
[9]. Purohit P, Dehury S, Patel S, Patel DK, Prevalence of Deletional Alpha Thalassemia and Sickle Gene in a Tribal Dominated Malaria Endemic Area of Eastern IndiaISRN Haematology 2014 2014ID 745245 [Google Scholar]