A Challenging Diagnosis of Fluctuating Dyspnea: Myasthenia Gravis
Rohan Mandaliya1, Kandan Kulandaivel2, Nicholas Nowotarski3, Kajalben Buddhdev4, Rajesh Patel5
1Resident, Department of Medicine, Abington Memorial Hospital, 1200 old York Road, Abington, PA, USA.
2Physician, Department of Neurology, Abington Memorial Hospital, 1200 old York Road, Abington, PA, USA.
3Medical Student, Temple University School of Medicine, 3500 N Broad St, Philadelphia, PA, USA.
4Resident, Department of Medicine, Abington Memorial Hospital, 1200 old York Road, Abington, PA, USA.
5Physician, Department of Pulmonary and Critical Care, Abington Memorial Hospital, 1200 old York Road, Abington, PA, USA.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Rohan Mandaliya, Resident, Department of Medicine, Abington Memorial Hospital, 1200 old York Road, Abington, PA-19001, USA.
E-mail: Rohan86m@gmail.com
Isolated respiratory muscles weakness is an unusual presentation of Myasthenia Gravis (MG). We present a case of 49-year-old male with undiagnosed MG who presented with fluctuating dyspnea on exertion and at rest. There was no peripheral muscle involvement, diagnostic tests and standard electromyography was normal, and the serologic tests were negative. The repetitive nerve stimulation and Single Fiber Electromyography revealed the diagnosis of MG. Interestingly the patient did not have sufficient therapeutic response on first line agents and required immunoglobulins and corticosteroids due to respiratory muscle involvement. Since skeletal muscle weakness is a cardinal feature of MG, dyspnea may be fluctuating as in our patient causing further difficulty in diagnosis as the pulmonary function tests performed during remission may be normal. This presentation highlights the need to consider MG in the differential diagnosis of unexplained dyspnea on exertion/rest.
Muscle weakness, Respiratory muscles weakness, Single fiber electromyography
Case Report
A 49-year-old male with a history of hypertension well controlled with a thiazide, was admitted to the hospital with a one month history of dyspnea on exertion which had progressed at rest. He became more dyspneic on immersing/standing in a swimming pool with water at the level of chest.
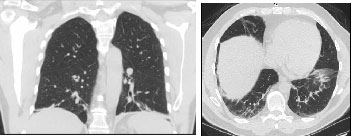
On examination the patient appeared exhausted and was not able to complete a full sentence without a pause. Respiratory rate was 26/min with an oxygen saturation of 94% on room air which dropped to 90% upon speaking. Physical examination was otherwise normal. Arterial blood gas revealed primary respiratory alkalosis. Complete blood count and Comprehensive Metabolic Profile were normal. Chest x-ray revealed mild interstitial opacities at the lung bases which were confirmed on CT scan of the chest [Table/Fig-1,2]. Cardiac brain natriuretic peptide and echocardiography were normal. Salicylate levels were normal. Evaluation of interstitial lung disease including urine legionella antigen, HIV antibody, ANA titer, hypersensitivity panel and fungal serologies were all negative.
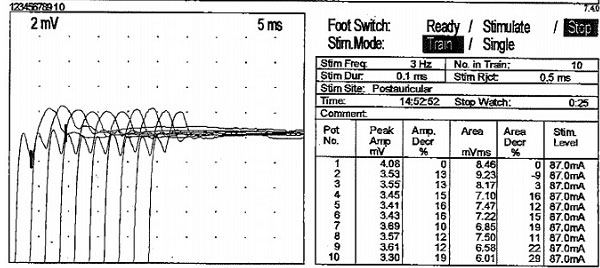
The patient’s dyspnea resolved with rest and supplemental oxygen. The patient was discharged, but re-admitted in two weeks with recurrence of his symptoms. Repeat CT scan of the chest showed no interval change. His symptoms again resolved with rest and oxygen. Due to the fact that his symptoms were relieved at rest and fluctuating, a neurologic etiology was considered. MRI of the brain was normal. Inspiratory and expiratory chest radiographs during his recovery phase were normal. The patient was examined by two different experienced neurologists with unremarkable neurological examination. The patient was discharged with close Pulmonary and Neurology follow-up. Outpatient Pulmonary Function Tests were normal: FVC 4.40L (96%); FEV1 3.30L (89%); FEV1/FVC 75%. TLC 7.42(112%); RV 3.02 (144%); DLCO 98%. Maximal inspiratory pressure, maximal expiratory pressure, vital capacity and 6 min walk test were also normal. Serologic tests for Acetylcholine receptor (blocking and modulating antibodies), Muscle Specific Kinase, Voltage gated calcium channel and paraneoplastic antibodies were negative. Electro-diagnostic studies revealed a normal needle electromyography. The absence of denervation on needle EMG ruled out Amyotrophic lateral sclerosis. Slow repetitive stimulation (at a rate of 3 Hz) of the right facial nerve after exercise, revealed greater than 10% decrement of the compound action muscle potential [Table/Fig-3]. Single Fiber Electromyography revealed increased jitter with occasional conduction block. These findings were consistent with Myasthenia Gravis.
The patient was started on a high dose pyridostigmine (60 mg five times per day), with improvement in his dyspnea and exhaustion. After a month his dyspnea was getting worse, so he was admitted to the hospital and was treated with oral prednisone (60 mg/day) and intravenous immunoglobulins (2 gm/kg; total dose) for five days along with home dose of pyridostigmine. His symptoms markedly improved. Eventually after two months he had involvement of other skeletal muscles causing neck extension weakness and diplopia. During the next six months the patient had been admitted twice for immunoglobulin therapy for worsening respiratory failure symptoms. The patient was in remission on chronic maintenance prednisone of 40 mg per day and pyridostigmine 60 mg five times a day thereafter for two months. However, since he was experiencing weight gain, swelling and mood changes, corticosteroids were stopped. Azathioprine was recommended for maintenance of remission, however the patient did not consider at that point because of its adverse effects. At present he is maintained in remission with pyridostigmine 60 mg five times a day and he would receive intravenous immunoglobulins if needed for worsening symptoms.
Discussion
This clinical presentation demonstrates several important learning points. It is important to consider neurological causes when dyspnea is unexplained by cardiac and respiratory causes. Neurological causes of dyspnea can be overlooked especially when they present with isolated respiratory muscle involvement as in our patient. Myasthenia Gravis (MG), a disease of the neuromuscular junction, is often complicated by respiratory failure known as myasthenic crisis; however most of the patients do so during the late course of the disease and already have other neurologic symptoms and signs. The presentation and progression of MG varies. Majority of the patients are female. More than half of the patients present with ocular symptoms (ptosis/diplopia), about 15% present with bulbar symptoms (dysarthria/dysphagia) with about 5% with proximal limb weakness [1].
However, MG can rarely present as isolated respiratory muscle weakness making the diagnosis extremely challenging. So far only four cases have been reported in the literature with respiratory muscle weakness as the initial presentation of MG without any Gravisother muscle involvement. [Table/Fig-4] shows the characteristics of the patients reported in the literature and the present patient with isolated respiratory muscle weakness as the initial presentation of MG [2-5].
Fluctuating skeletal muscle weakness is a cardinal feature of MG [6]. Characteristically, muscular exertion increases the myasthenic weakness. Thus, dyspnea may be fluctuating as in our patient causing further difficulty in diagnosis as the pulmonary function tests performed during remission may be normal. Interestingly the patient had dyspnea on immersion which can be specific for neuromuscular diseases [7]. It is likely due to diaphragmatic weakness affecting vital capacity and gas exchange. Our patient had basilar infiltrates on chest radiographs and CT scan that likely represented atelectasis. Patients with MG involving respiratory muscles frequently have atelectasis due to decreased diaphragmatic and respiratory muscle movement.
The diagnosis of MG can be challenging in certain group of patients. It is important to note that serologic testing with Ach receptor antibodies can be negative in 10-20% of the patients [6]. Our patient did not have positive serologies for acetylcholine receptor (Ach receptor). Some of these seronegative patients have low affinity antibodies to AChR that cannot be detected in standard solution phase assays. A British group devised a novel laboratory method and detected AChR antibodies in two thirds of sera previously negative for binding to AChR in solution [7]. However, it has not been introduced commercially yet.
In a subset of seronegative patients, myasthenic weakness comes from autoimmune process directed to postsynaptic targets distinct from the AChR [8]. Antibodies against muscle specific kinase are present in about 40% of seronegative cases with generalized MG, and in another portion of myasthenic patients, antibodies against lipoprotein receptor protein (LRP4) are detectable [9]. MuSK and LRP4 are not involved directly in the neuromuscular transmission, but in the end plate maturation. Patients can still be triple seronegative. Based on this, a new subgroup of MG called, “Seronegative MG” has been recently included in the classification of MG [9].
The two electrophysiologic tests used for the diagnosis of MG are repetitive nerve stimulation test and single fiber electromyography. Repetitive nerve stimulation alone is positive in 75% of the cases in generalized MG and 50% of the cases in ocular MG. Single fiber electromyography is the most sensitive test for diagnosis of MG (95%-99%); available at few centers only. Patients with MG will have varying onset of action potentials due to abnormal neuromuscular transmission and, when the recordings are superimposed, there is variability in the onset of action potentials producing “jitter” in the recordings.
CT scan of the chest in coronal and transverse views respectively shows bilateral interstitial reticulonodular opacities at lung bases
Slow repetitive stimulation (at a rate of 3 Hz) of the right facial nerve after exercise, reveals greater than 10% decrement of the compound action muscle potential in almost all stimulus
Comparison of patients reported in the literature with isolated respiratory muscle weakness as the initial presentation of Myasthenia Gravis
| Present Patient
| Patient 2 [2] | Patient 3 [3] | Patient 4 [4] | Patient 5 [5] |
|---|
| Age | 49 | 64 | 22 | 68 | 61 |
| Sex | M | M | F | F | F |
| Presenting symptom | Dyspnea on exertion progressed to rest | Dyspnea on exertion progressed to rest | Dyspnea on exertion progressed to rest | Dyspnea on exertion progressed to rest | Dyspnea on exertion progressed to at rest |
| EMG | Normal | Normal | Positive | Positive | Not performed |
| Serology | Normal | Positive | Positive | Positive | Positive |
| SFEMG | Positive | Not performed | Not performed | Not performed | Not performed |
| Enlarged thymus | No | No | No | No | Yes |
| Treatment | High dose Acetylcholinesterase inhibitors showed transient response. Immunoglobulins and steroids intermittently for respiratory failures with resolution of symptoms. | Acetylcholinesterase inhibitors and steroids not helpful. Plasma exchange helpful. | Acetylcholinesterase inhibitors, steroids and immunoglobulins due to severe respiratory failure with resolution of symptoms. | Acetylcholinesterase inhibitors, steroids and immunoglobulins due to severe respiratory failure with resolution of symptoms. | Acetylcholinesterase inhibitors, steroids and plasma exchange with resolution of symptoms. |
Conclusion
Thus Myasthenia Gravis should be considered in patients with unexplained dyspnea and its diagnosis be aggressively pursued in spite of negative serologies with single fiber electromyography being the most sensitive test.
[1]. D Grob, N Brunner, T Namba, M Pagala, Lifetime course of myasthenia gravisMuscle Nerve 2008 37:141-49. [Google Scholar]
[2]. KM Dushay, JD Zibrak, WA Jensen, Myasthenia gravis presenting as isolated respiratory failureChest 1990 97:232-34. [Google Scholar]
[3]. SR Sharma, N Sharma, M Yeolekar, An undiagnosed myasthenia gravis presenting as isolated recurrent acute respiratory failureJ Neurosci Rural Pract 2012 3:80-82. [Google Scholar]
[4]. WH Kim, JH Kim, EK Kim, SP Yun, KK Kim, WC Kim, Myasthenia gravis presenting as isolated respiratory failure: a case reportKorean J Intern Med 2010 25:101-04. [Google Scholar]
[5]. N Kishimoto, H Kondou, Myasthenia gravis presenting as isolated respiratory failureNihon Kokyuki Gakkai Zasshi 1998 36(10):891-95. [Google Scholar]
[6]. BM Conti-Fine, M Milani, HJ Kaminski, Myasthenia gravis: past, present, and futureJ Clin Invest. 2006 116(1):2843-54. [Google Scholar]
[7]. B Schoenhofer, D Koehler, MI Polkey, Influence of immersion in water on muscle function and breathing pattern in patients with severe diaphragm weaknessChest 2004 125:2069-74. [Google Scholar]
[8]. MI Leite, S Jacob, S Viegas, IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravisBrain 2008 131:1940-52. [Google Scholar]
[9]. JP Sieb, Myasthenia gravis: an update for the clinicianClin Exp Immunol 2014 175:408-18. [Google Scholar]